Computer Validation: Do All Roads Lead to Annex 11?
Special Issues
The European regulations on good manufacturing practices (GMPs) - Annex 11 - are having a strong influence on good laboratory practice (GLP) and good clinical practice (GCP).
Volume 29, Number 12
Pages 10-13
For computerized system validation, do all roads lead to Annex 11? The recently issued draft advisory document "Application of GLP [Good Laboratory Practice] Principles to Computerised Systems" from the Organisation for Economic Co-operation and Development (OECD) is heavily influenced by European Union Good Manufacturing Practice (EU GMP) Annex 11. This column explores the influence of a GMP regulation on GLP and good clinical practice (GCP) disciplines.
Some of the more printable comments that my children say when they see me reading health care regulations and guidance are "sad" and "get a life." However, when I began reading the new draft good laboratory practice (GLP) regulatory guidance for computerized systems issued by the Organisation for Economic Co-operation and Development (OECD) I noticed that there were similarities between it and European Union Good Manufacturing Practice (EU GMP) Annex 11. From this observation, I developed the idea for this column. Read on and see how a good manufacturing practice (GMP) regulation for computerized systems is taking over most of the regulated world (except in the United States).
In the Beginning
In the beginning, EU GMP Annex 11 (1) outlined the requirements for computerized systems in a GMP environment. Annex 11 was originally published in 1992 and was updated in 2011. Shortly after the latter version was published, I wrote a "Focus on Quality" column on this subject (2) explaining the main points of the new regulation. The scope of GMP covers pharmaceutical manufacturing quality control laboratories, quality assurance, and the production of new drugs called investigational medicinal products in Europe or investigational new drugs in the United States. Therefore, computerized systems in these functional areas, especially laboratory computerized systems, carrying out regulatory work have to follow these regulations in Europe and those countries that follow Pharmaceutical Inspection Convention/Scheme (PIC/S) GMP regulations (3). Under EU or PIC/S GMP the road is very clear and the regulations lead directly to Annex 11.
However, the title of this column also indicates that for the other good practice disciplines, for example, GLP and good clinical practice (GCP), the tentacles of Annex 11 reach out from GMP and into these areas. Figure 1 shows an overview of how Annex 11 impacts GLP, GCP, and computerized systems.

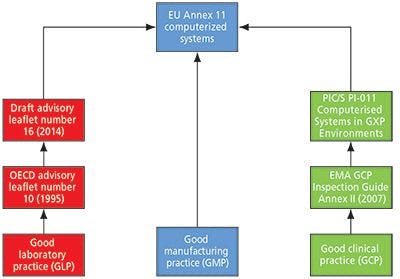
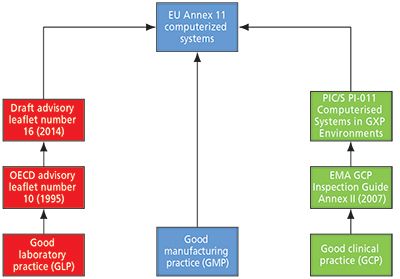
Figure 1: The influence of EU GMP Annex 11 on other good practice disciplines.
Good Laboratory Practice
In Europe, the applicable GLP regulations are published by the OECD, which is based in Paris, France (4). In 1995, the OECD published a consensus document on "The Application of the Principles of GLP to Computerised Systems" (5), which was a collaboration between inspectors and pharmaceutical industry experts.
However there are a number of deficiencies with this document:
- No mention of specification of the requirements for the system in the validation section (apart from the definition of validation in the introduction).
- No defined life cycle for a system validation.
- No definition of the expected documentation required to demonstrate validation.
The focus is more on the operational aspects of the system - for example, disaster recovery, change control, security, system description, and so on. The document is clearly showing its age now, so a draft replacement document was issued in September 2014 for industry comment (6). The draft document is entitled "The Application of GLP Principles of Computerised Systems" - please note the snappier title! After it has been reviewed, updated, and approved, it will replace the 1995 document.
What has changed? We now have a 23-page document that is relatively detailed when industry wants the ability to interpret regulations rather than be presented with a proscriptive shopping list. However, the main thing you'll notice when reading the draft is how heavily influenced it is by Annex 11. For example, in clause 12 the wording of the first two sentences is "Risk assessment should be applied throughout the life cycle of a computerised system taking into account data integrity and the quality of the study results. Decisions on the extent of validation and data integrity controls should be based on a justified and documented risk assessment." Apart from the modification of the wording for a GLP study and dropping the requirements for product quality and patient safety, the intent and purpose of this wording is the same as in Annex 11.
Electronic signature requirements outlined in clause 138 are shamelessly plagiarized virtually word for word from Annex 11 with the substitution of "test facility" for "company" and "legal consequences" for "impact." As that great computer validation expert Oscar Wilde said, "Plagiarism is the sincerest form of flattery." When something works don't change it. However, where Annex 11 defines requirements for electronic signatures in 40 words and leaves it to a company to interpret the regulation, the draft OECD document then meanders on with another eight clauses of additional requirements for electronic signatures over a page of text. These additional requirements are essentially an unnecessary interpretation of clause 138. This is where the document becomes overbearing.
You can now understand the GLP section on the left-hand side of the flow chart in Figure 1. The draft OECD document is Annex 11 that has been expanded and modified for GLP. Thus, the GLP computer validation road leads to Annex 11.
I can't resist some further comments about the draft OECD document because it is so new. There are some good clauses in the document:
- The requirement for dual-use systems (GLP and non-GLP) to be subject to validation in clause 14 is a good approach.
- Test facility management has overall responsibility for validation, and this is outlined in the section on roles and responsibilities (clauses 15–24 plus a table at the end of the document).
- The need for user requirements specification (URS) for retrospective validation is good (clause 67). Although the update to Annex 11 withdrew the ability for retrospective validation, I think the final version of this document should do the same. Organizations have had enough time to validate legacy systems since 1995 - why permit them any further slack?
However, there are also a number of clauses that have no place in a modern regulatory document dealing with computerized systems. Here are a few comments about just a small selection of those clauses:
- The organization of the document is appalling. Why is the URS in the operational phase, but not under the project phase for validation? Most of the operational stuff appears in the project phase.
- The scope of the document includes the validation of balances (clause 9). What?! Have the people who wrote this document taken leave of their senses or taken illicit substances? We qualify balances and we don't validate them.
- Clause 77 on testing brings the competence and knowledge of the writers of this document into question. It states "Qualification testing e.g. Design Qualification . . . ." Design qualification is a way of specifying requirements under USP <1058> (7) or in some other computer system validation (CSV) systems - it is the confirmation that the selected system meets the user requirements. Under no circumstances is it testing. It would be better to simply state that software needs to be properly installed and tested against the requirements in the URS.
- Clauses 47 and 75 talk about availability of source code for inspectors. I would be very interested in knowing what education, training, and experience inspectors have to review this material.
- Raw data and derived data are mentioned in clauses 97 and 98, but associated metadata are not considered until clause 105. The principle of a data trail from the original observations to the study report is good, but the material needs to collated into a cogent approach.
- Clause 122: Performance assessment seems like a waste of time as the IT department should, for networked systems, have performance monitoring software in place to determine how well a system operates on a continuous basis.
Overall, this document needs a lot of work and I suspect that industry feedback will not be positive. That said, it is an advance on the existing guidance but not as far forward as the authors would have hoped.
Good Clinical Practice
The link between GCP and the regulations for computerized systems in Annex 11 may seem obscure, but the connection is shown on the right hand side of Figure 1. In Europe, the European Medicines Agency (EMA) has issued a number of GCP inspection guidance documents for carrying out inspections of clinical sites, investigators, and sponsors. Appendix III deals with computerized systems and is one page in length (8). It simply states that "The EU GCP inspectors agreed to use as the reference for inspection of Computer Systems the published PIC/S Guidance on Good Practices for Computerised Systems in Regulated 'GXP' Environments (PI 011-3)."
Therefore, GCP inspections will use a document based on GMP for their inspection of computerized systems in GCP. The document PI-011-3 (9) is dated 2007, but the text was written around 2002 and is based on GAMP 4 guidance (10) rather than the most recent version, GAMP 5 (11). The two subsequent updates are merely corrections of errors and an address change, which subsequently hide the document's actual age.
You may be asking, how does this document lead to Annex 11? The answer lies in the next version of the guidance as the team of inspectors that updated Annex 11 and Chapter 4 are currently involved in updating this document. The current version of PI-011 does not lead directly to Annex 11, but the revised version most certainly will.
Quo Vadis FDA?
So far this column has focused on regulations outside of the United States, mainly discussing the EU and OECD whose regulations are relatively easy to update. For example, by March 2015, eight of the nine chapters of EU GMP will have been updated since 2011. In contrast, it appears relatively difficult for the Food and Drug Administration (FDA) to change regulations, for example, 21 CFR 11 (12) in 1997 and the minor changes to 21 CFR 211 (13) in 2008. Most of the recent "regulations" issued by the FDA comes in two forms either Level 1 guidance, where the agency issues formal guidance for industry documents or Level 2 guidance that are posted in the FDA web site.
The two main Level 1 guidance documents for computerized systems are the "General Principles of Software Engineering" (14) and "Computerized Systems in Clinical Investigations" (15). The problem with the former document is that it is written primarily for the development of medical device software that cannot be configured or customized by a user. Therefore, the project phase for implementation that would typically include configuration of software to meet a business process is missing. The latter guidance document is focused only on the operational and compliance requirements when conducting clinical trials using a computerized system. Neither document provides holistic guidance for validating a computerized system and maintaining the validation status in a regulated laboratory.
Level 2 guidance is posted on the agency's web site and can be useful. Under the GMP guidance (16) the following questions are posed (and answered):
- 3. How do the Part 11 regulations and "predicate rule requirements" (in 21 CFR Part 211) apply to the electronic records created by computerized laboratory systems and the associated printed chromatograms that are used in drug manufacturing and testing? (published 2010)
- 5. Why is FDA concerned with the use of shared login accounts for computer systems? (published 2014)
- 7. In warning letters to firms, why has FDA objected to the practice of using actual samples to perform system suitability testing (sometimes also referred to as "trial," "test," or "prep" runs)? (published 2014)
The answers are useful, but you need to think wider than the published question and answer session. For example, if you looked at question 3 and just thought the answer focused solely on chromatographic data you would be wrong. It covers all data generated in the laboratory by all computerized systems. Whichever way you look at it, there is a dearth of FDA regulatory guidance for computerized system validation. Thus, from the perspective of Annex 11 the road has hit a dead end.
Summary
There appears to be a de facto harmonization within Europe that regulations or regulatory guidance is coalescing around EU GMP Annex 11. We do not know if this is by design or serendipity, but from the actual evidence and direction of travel, both shown in Figure 1. However, Annex 11 is the closest thing to regulatory harmonization for computerized system validation. Further information on the global applicability of Annex 11 in GMP can be found in a book edited by Orlando Lopez to be published in early 2015 (17).
References
(1) European Commission Health and Consumers Directorate-General, EudraLex: The Rules Governing Medicinal Products in the European Union. Volume 4, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. Annex 11: Computerised Systems (Brussels, Belgium, 2011).
(2) R.D. McDowall, Spectroscopy26(4), 24–33 (2011).
(3) Pharmaceutical Inspection Convention/Scheme, available at www.picscheme.org (PIC/S, Geneva, Switzerland).
(4) Organisation for Economic Co-operation and Development (OECD) Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 1, OECD Principles on Good Laboratory Practice, (OECD, Paris, France, 1998).
(5) Organisation for Economic Co-operation and Development (OECD) Series on Principles of Good Laboratory Practice and Compliance Monitoring Number 10, The Application of the Principles of GLP to Computerised Systems, (OECD, Paris, France 1995).
(6) Organisation for Economic Co-operation and Development (OECD) Series on Principles of Good Laboratory Practice and Compliance Monitoring Draft Number 16, The Application of GLP Principles of Computerised Systems, (OECD, Paris, France, 2014).
(7) United States Pharmacopeia General Chapter <1058> "Analytical Instrument Qualification" (United States Pharmacopeial Convention, Rockville, Maryland).
(8) "Procedure for Conducting GCP Inspections Requested by the EMEA: Annex III Computer Systems," (European Medicines Agency, 2007).
(9) Pharmaceutical Inspection Convention/Scheme, "Computerised Systems in GXP Environments (PI-011-3)" (PIC/S, Geneva, Switzerland 2007).
(10) ISPE, Good Automated Manufacturing Practice (GAMP) Guide Version 4 (International Society of Pharmaceutical Engineers, Tampa, Florida, 2001).
(11) ISPE, Good Automated Manufacturing Practice (GAMP) Guide Version 5 (International Society of Pharmaceutical Engineers, Tampa, Florida, 2008).
(12) Electronic Records; Electronic Signatures, Final Rule, 21 CFR 11 (U.S. Government Printing Office, Washington, DC, 1997)
(13) Current Good Manufacturing Practice for Finished Pharmaceutical Products, 21 CFR clause 211 (U.S. Government Printing Office, Washington, DC, 2008).
(14) US Food and Drug Administration, Guidance for Industry, General Principles for Software Validation (FDA, Rockville, Maryland, 2002).
(15) US Food and Drug Administration, Guidance for Industry, Computerized Systems in Clinical Investigations (FDA, Rockville, Maryland, 2008).
(16) US Food and Drug Administration, "Questions and Answers on Current Good Manufacturing Practices, Good Guidance Practices," Level 2 Guidance - Records and Reports, FDA, Rockville, Maryland, http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm).
(17) EU Annex 11 Guide to Computer Validation Compliance for the Worldwide Health Agency GMP, O. Lopez, Ed. (CRC Press, 2015, http://www.crcpress.com/product/isbn/9781482243628).
R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com



R.D. McDowall























