High-Resolution Native Mass Spectrometry Opens the Door for Detailed Analyses of Intact Protein Complexes
Native mass spectrometry, the method by which noncovalent protein complexes are retained in the gas phase for intact mass analysis, is gaining interest as a method for intact protein characterization. The development of a modified orbital ion trap platform for high-resolution analyses has expanded the role of native mass spectrometry to address the challenges of intact protein characterization.
Native mass spectrometry, the method by which noncovalent protein complexes are retained in the gas phase for intact mass analysis, is gaining interest as a method for intact protein characterization. The development of a modified orbital ion trap platform for high-resolution analyses has expanded the role of native mass spectrometry to address the challenges of intact protein characterization. Here, we give an overview of the types of analyses that have recently been accomplished with this new instrumentation, ranging from highly decorated small proteins to large, noncovalent complexes that bind small-molecule ligands. These examples highlight the capabilities of native mass spectrometry and hint at the future role in intact protein characterization.
The importance of protein analysis has expanded as the role that proteins play in disease progression and potential treatment has become increasingly evident. Many of the diseases we seek to treat stem from phenomena such as misregulation of proteins or misfolding of protein structure. As a way to combat these types of diseases, there is a push for the use of proteins as therapeutics, most notably the development of disease-specific antibodies (1,2). As an example, monoclonal antibodies (mAbs) have been developed to target human epidermal growth factor receptor 2 (HER2) and tumor necrosis factor (TNF) to treat cancers, rheumatoid arthritis, and Crohn's disease. These antibodies number in the top-selling therapeutics (3). To understand these diseases and develop protein-based treatments, it is necessary to delve into protein structure and interactions to construct a detailed network.
Advances in technology have aided in the characterization of proteins and protein networks. The determination of protein primary sequence is currently routine in many laboratories, but it is the characterization of protein–protein interactions and post-translational modifications (PTMs) that remains challenging. PTM analysis is especially crucial because cellular signals are often transduced through PTMs, such as phosphorylation, and PTMs can result from drug degradation (for example, oxidation). Also, for therapeutic antibodies, glycosylation plays a key role in drug clearance and potential immunogenicity. There are several analytical techniques for studying protein interactions and PTMs, such as ion-exchange chromatography (IEC), enzyme-linked immunosorbent assay (ELISA), capillary electrophoresis (CE), and native polyacrylamide gel electrophoresis (PAGE), but mass spectrometry (MS) proteomics methods have become the workhorse for many of these analyses. Often, these interactions and PTMs are detected using enzymatic digestion of a cell lysis or pull-down followed by separation and characterization of the peptides (4). However, this process does not reveal the nature of the intact complexes, and thus these interactions can be hypothesized but not confirmed. Analysis of intact proteins and protein complexes would not only confirm the identity of cellular protein complexes but also provide insight to the multiple proteoforms present (5).
MS has become a crucial technology for the characterization of proteins and protein complexes as advances make it more sensitive and robust. MS was initially relegated to the realm of small-molecule analysis because of the inability to transfer proteins to the gas phase efficiently and effectively. Development of "soft" ionization techniques such as electrospray ionization (ESI) broadened the utility of MS by allowing intact proteins to be detected (6). Though the intact protein was then retained in the gas phase, the protein structure was often lost because of the organic solvents and acids used as the ionization solution. The development of nano-ESI, which uses a smaller orifice and lower ionization voltages, allowed the incorporation of buffers consisting of aqueous volatile salts, such as ammonium acetate, as ionization solutions, so that aspects of protein higher-order structure and noncovalent interactions could be retained in the gas phase. This native MS (7) is applicable for a wide array of protein samples, with complexes up to 18 MDa (8,9). Recent developments in MS instrumentation have led to the incorporation of native MS conditions on an orbital ion trap platform (10). This combination has yielded high-resolution analyses of proteins and protein complexes, with the ability to characterize complex, heterogeneous PTMs on an intact level. Here, we highlight some recent applications probed using this technology. These examples span the range of size currently achievable as well as emphasize the ability to characterize multiple proteoforms simultaneously present in a single sample. These applications demonstrate the utility of native MS to provide thorough characterization of heterogeneous mixtures and a basis for the future role of native MS for intact protein analysis.
Methods
Below is a general summary of the sample preparation, instrumentation, and data analysis methods used for native MS of intact proteins and protein complexes. For precise details of these protocols, we direct you to several recent reviews (11,12).
Sample Preparation
A clear advantage of native MS is the simplicity of sample preparation necessary for analysis. The success of native MS hinges on the use of volatile aqueous buffers as solutions for nano-ESI, thus allowing the retention of noncovalent interactions into the gas phase. For the most part, pH-neutral ammonium acetate solutions are used. Protein samples, which are typically purified, are buffer-exchanged using molecular weight cutoff (MWCO) spin filters to ammonium acetate solutions of similar ionic strength and pH as the initial buffer. It is possible to treat the sample before buffer exchange to reduce the complexity of the proteoforms using an enzyme such as a glycosidase or phosphatase. Precise sample treatment depends on the goal of the experiment and will be sample dependent. After buffer exchange, 1–2 µL of a 1–10 µM sample solution is typically introduced to the mass spectrometer under static flow conditions using gold-coated borosilicate capillaries generated in-house.
Instrumentation
Because native MS uses the "gentle" ionization conditions of aqueous volatile buffer and nano-ESI, fewer charges are imparted to the protein ion, thus shifting the charge state distribution to higher m/z values. Until recently, this shift meant that time-of-flight (TOF) mass analyzers were required because they were able to provide the mass range necessary for detection. A few instrument modifications are necessary to ensure efficient desolvation, transmission, and detection of these large ions. To assist in desolvation and transmission, the pressure in the first vacuum state is increased (13–15). Other modifications include the incorporation of a high transmission grid and low rep rate pusher in the TOF system, as well as the potential use of a low-rf quadrupole and high-pressure collision cell of tandem MS experiments (16,17).
As mentioned previously, technological developments have made it such that native MS is now possible on an orbital ion trap platform (10). Native MS was initially incorporated on a modified Exactive Plus instrument (Thermo Fisher Scientific). The modifications include manipulations of rf voltages applied to the transfer multipoles to improve ion transmission, software alterations to allow detection of higher m/z ions, and manual control of gas composition and pressure in the collision-induced dissociation cell. With these modifications, it is possible to scan from 400 to 30,000 m/z and achieve a mass resolution of 25,000 at m/z 5000 and 16,000 at m/z 10,000, as determined by cesium iodide (CsI) clusters used for mass calibration.
Data Analysis
Deconvolution of the raw data was performed using Protein Deconvolution 2.0 software (Thermo Fisher Scientific). From the zero-charge spectrum, the accurate masses were obtained and the peak intensity was used for any relative quantitation.
Recent Applications
As mentioned previously, native MS is applicable for the analysis of a wide array of intact proteins and protein complexes; however, the achievable mass resolution remained an issue because of inefficient desolvation. The use of native MS on an orbital ion trap platform addressed this challenge and, now, highly detailed analyses are possible. Below, we present examples that illustrate the level of detail that is achievable in intact protein analysis.



Figure 1: High-resolution native MS identifies 59 proteoforms of intact chicken ovalbumin. The glycan heterogeneity was most easily determined using the dephosphorylated form (top spectrum in black) and each glycan is indicated by a pink circle. Analysis of unprocessed ovalbumin (bottom spectrum in blue) allowed identification of multiple phosphorylation sites, indicated by blue stars. The high mass signals (gray box, multiplied by a factor of 10) are relatively low in abundance yet significantly consist of the less-reported glycan structures. Adapted from reference 18 (copyright ACS Publications).
Defining Protein Microheterogeneity
PTMs play an important role in maintaining the structure and regulating the function of proteins. The detection and analysis of PTMs has progressed with advances in technology, leading to protocols that enrich for PTM-labeled peptides or analysis of released glycans and glycopeptides, for example. Because of the complexity of the microheterogeneity of proteins, PTM analysis of intact proteins remains a challenge. Most of these methods require reduction of sample complexity, often through enzymatic digestion of the protein backbone. In this way, the complete picture of co-occurring PTMs can be lost. The improvements in resolution obtained via the combination of native MS on an orbital ion trap system allow more complete analysis of this microheterogeneity.



Figure 2: Glycan analysis of intact hingeless IgG4 antibodies. (a) The lack of the covalent disulfide bonds that hold the heavy chains together results in the simultaneous analysis of the half- and whole IgG4 antibody. The high resolution achieved (insets, a) allowed analysis of the glycan heterogeneity (b and c). The deconvoluted spectra show the identification of glycans typically observed for intact antibodies for both (b) the half-body and (c) the whole antibody. Adapted from reference 24 (copyright Landes Bioscience, Inc.).
The first example to highlight these improvements is the complete characterization of ovalbumin (18). Ovalbumin is a relatively small protein (approximately 45 kDa) that is decorated with an abundance of PTMs, including phosphorylation, glycosylation, and a disulfide bridge (19,20). The combination of these different PTMs has made intact protein analysis a challenge. We took on this challenge and demonstrated that this microheterogeneity could be characterized by native MS (Figure 1) (18). We identified 59 separate proteoforms, and more than half of the proteoforms presented at less than 5% relative abundance. These co-occurring low-abundance proteoforms are often lost using traditional methods. Complex, extensive glycosylation contributes to the sheer number of proteoforms, and multiple glycans were detected by native MS. Previous analyses of glycans from ovalbumin often attributed unexpected glycans to contaminant proteins (21,22). However, by performing PTM analysis on intact ovalbumin, the observed glycans can be confirmed as bound to ovalbumin, thus eliminating any uncertainty about their origin. The nature of the PTMs was confirmed by selective application of enzymes, such as a glycosidase or phosphatase, demonstrating that intact analysis by native MS was capable of providing a complete overview of the microheterogeneity of ovalbumin.



Figure 3: High-resolution native MS yields simultaneous characterization of drug-to-antibody ratio and glycan heterogeneity of antibodyâdrug conjugate. The commercially available cysteine-linked antibodyâdrug conjugate brentuximab vedotin (Adcetris) was analyzed in its (a) deglycosylated and (b) glycosylated forms. Adapted from reference 24 (copyright Landes Bioscience, Inc.).
Aside from complete characterization of a small heterogeneous protein, native MS can also be used to provide an overview of simpler PTMs on larger proteins and protein complexes, such as intact antibodies (mass of approximately 150 kDa). The utility of native MS has been demonstrated for glycan monitoring of half and whole IgG4 antibodies as well as an antibody–drug conjugate (23,24). Because of mutations in the IgG4 antibodies, the hinge was removed, and the two heavy chains were associated solely by noncovalent interactions. And, even though the stability of the antibody structure was reduced, the two heavy chains remained associated in the gas phase and appeared with sufficient resolution as to characterize the attached glycans (Figure 2) (24). In a similar manner, native MS yielded characterization of an antibody–drug conjugate, including both the drug-to-antibody ratio of the different species present and the attached glycans (Figure 3) (24). The use of native MS allowed the retention of noncovalent interactions yielding the intact antibody–drug conjugate even at a drug-to-antibody ratio of 8 (meaning that all disulfide bonds had been disrupted and linked to the cytotoxic drug). Glycoforms could readily be assessed because of clear separation of the signals corresponding to different drug-to-antibody ratios and baseline resolution between adjacent glycoforms. In some of the IgG4 half-antibodies studied, the glycan heterogeneity expanded drastically, with an increase in the branching as well as incorporation of sialic acid. Glycans containing sialic acid are typically a challenge to characterize when released from the protein backbone because of inefficient ionization (25,26). By analyzing the intact protein, the differences in ionization efficiency for the various glycans is reduced and quantitation is possible. This is evidenced through the quantitative comparison of the glycoforms before and after the use of a sialidase to clip the sialic acid from the glycans (24). Removal of sialic acid simplified microheterogeneity of the IgG4 half-antibody and changed the relative abundance of the glycoforms, but the differences in abundance can all be accounted by the simplification glycan structure. These examples emphasize the ability of native MS to retain noncovalent interactions with enough mass resolution that the glycan profiles of intact antibodies are observed, an aspect that could potentially be implemented as a screening technology.

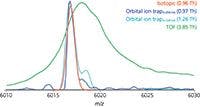

Figure 4: Experimental data approaches theoretical peak width. The overlap of raw native MS signals for a single IgG1 antibody is shown with that of the theoretical signal. The peak widths at half-maximum abundance (FWHM) is shown in parentheses. Adapted from reference 33 (copyright Landes Bioscience, Inc.)
Probing Mixture Composition
The previous examples demonstrate how native MS on an orbital ion trap platform can provide a detailed overview of the variety of PTMs that can be present on an intact purified protein. However, some samples are more complicated such as antibodies engineered to work as a mixture. As the level of bioengineering increases for antibody-based therapeutics, the analytical methods must also advance to tackle the challenges of characterizing a mixture of highly related components. For example, the production of antibody–drug conjugates often results in a heterogeneous mixture because of different drug payloads on the protein backbone. This is evident by the presence of multiple species differing by drug-to-antibody ratio (Figure 3) (24). Another example is the recent interest in using a combination of antibodies as a single therapeutic (2,27,28). Currently, these therapeutics consist of individual antibodies that are characterized individually and administered simultaneously (29). For this simple mixture, the analytical characterization methods are already established, but as mixtures become more complex, the cost of time and resources for the production of these mixtures grows to the point of being prohibitive. To address this future issue, there is a push for novel technology to produce mixtures of antibodies on a single platform (29–31). With these advances in mixture generation, analytical characterization becomes more difficult. In 2012, we compared native MS with cation-exchange chromatography for the analysis of these types of antibody mixtures and found that native MS yielded comparable quantitation with the advantage of identification and quantitation in a single experiment (32). We also noted that native MS is able to handle increasing mixture complexity better than cation-exchange chromatography. The increased resolution obtained via the orbital ion trap platform only expands the utility of native MS for mixture characterization. We recently probed the limits of this technique by characterizing mixtures containing 6, 10, and 15 antibodies (33). We found that baseline resolution was achieved with mass differences as little as 42.27 Da for intact, deglycosylated antibodies. A comparison of peak widths revealed that experimental resolution approaches that of the theoretical isotopic peak width resulting in supreme mass accuracy (Figure 4) (33). This mass accuracy, an average of 7 ppm, is crucial for consistent identification and confirming the primary sequence and PTMs. The mass accuracy, combined with high mass and quantitative precision, less than 7.5 ppm and 1.2%, respectively (Table I), forms the basis of a powerful method for comprehensive mixture characterization.

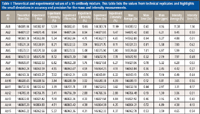
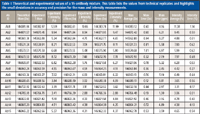
Table I: Theoretical and experimental values of a 15-antibody mixture. This table lists the values from technical replicates and highlights the small deviations in accuracy and precision for the mass and intensity measurements.
Obtaining Details for Protein Complexes
Analysis of single proteins is necessary for probing the complex microheterogeneity that is often present, but proteins rarely exist individually. Instead, protein complexes and the interactions of various subunits regulate functions and pathways. In the initial study of native MS on an orbital ion trap platform, a wide range of protein complexes was studied, spanning from intact antibodies (150 kDa) to the molecular chaperone GroEL (801 kDa) (10). Not only was baseline charge-state resolution achieved, but the addition of small molecules could be monitored. For example, incubation of GroEL with ADP or ATP resulted in the sequential binding of these small ligands to monomers in the 14mer protein complex. The addition of ADP or ATP molecules to intact GroEL was easily resolved (Figure 5), equating to additions of less than 0.1% total mass (10).

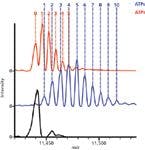
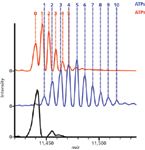
Figure 5: Native MS on the orbital ion trap platform allows investigation of small mass changes to high mass complexes. Detection of small molecule ligands binding to large intact protein complexes is illustrated by the binding of ADP and ATP to intact GroEL (801 kDa). The mass spectra show unbound GroEL (black) and that bound to ADP (red) or ATP (blue). The baseline resolution between adjacent species allows the counting of individual molecules, as indicated by the numbers. Adapted from reference 10 (copyright Nature Publishing Group).
Discussion and Future Outlook
The drive for development of novel biotherapeutics also pushes the limits of traditional analytical technologies leading to the concurrent development of comprehensive new methods. Native MS has been gaining interest as a result of its many applications, like those described above. The combination of native MS on an orbital ion trap platform has expanded its utility to now be able to provide a detailed overview of the microheterogeneity of intact proteins and protein complexes. Several other advantages make native MS a valuable asset for biotherapeutic characterization. Inherent to MS-based techniques, very little sample is required (on the order of femtomoles) and, unique to native MS, uses a straightforward, limited sample preparation without worrying about the reproducibility of proteolytic digestion. The rapid acquisition time means that results are obtained in minutes compared to hours and copurified contaminant proteins are often evident because of drastic differences in mass. However, these analyses do require specialized instrumentation that is often modified to transmit and detect these large ions (see above). While native MS provides a comprehensive overview of protein microheterogeneity, exact localization and structure of the PTMs, such as the connectivity and branching of a glycan, cannot be determined. Also, several PTMs result in moderate to large differences in mass, but there are those like deamidation that are much more readily detected by cation-exchange chromatography than native MS.
The high resolution afforded by the combination of native MS on an orbital ion trap platform can provide the detailed analysis of intact proteins that may be used as a high-resolution fingerprint for analytical characterization of biotherapeutics. Native MS has the ability to identify various components in a mixture, whether they be PTM-based microheterogeneity or highly related antibodies in a composite mixture, as well as quantitate these components in a single experiment. The retention of noncovalent interactions and tertiary structure allows the creation of a complete picture of the intact protein and its proteoforms as well as expanding the applicability to protein complexes. Improvements in automation, both in sampling (34,35) and data analysis (36,37), are leading to simplifying the implementation of this technology in the laboratory. In our opinion, the benefits of high-resolution native MS would be readily realized in the characterization of complex biotherapeutics and can provide insight to the microheterogeneity of protein structure and function.
Natalie J. Thompson and Albert J.R. Heck are with the Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences at Utrecht University in Utrecht, The Netherlands as well as the Netherlands Proteomics Centre. Direct correspondence to: a.j.r.heck@uu.nl
References
(1) J.M. Reichert and E. Dhimolea, Drug Discovery Today 17, 954–963 (2012).
(2) T. Robak, Expert Opin. Biol. Ther. 13, 953–958 (2013).
(3) I. Strickland, EvaluatePharma 1–38 (2012).
(4) A.F. Altelaar, J. Munoz, and A.J. Heck, Nat. Rev. Genet. 14, 35–48 (2013).
(5) L.M. Smith and N.L. Kelleher, Nat. Methods 10, 186–187 (2013).
(6) J.B. Fenn, M. Mann, C.K. Meng, S.F. Wong, and C.M. Whitehouse, Science 246, 64–71 (1989).
(7) J.A. Loo, Int. J. Mass Spectrom. 200, 175–186 (2000).
(8) A.J. Heck, Nat. Methods 5, 927–933 (2008).
(9) J. Snijder, R.J. Rose, D. Veesler, J.E. Johnson, and A.J. Heck, Angew. Chem. 52, 4020–4023 (2013).
(10) R.J. Rose, E. Damoc, E. Denisov, A. Makarov, and A J. Heck, Nat. Methods 9, 1084–1086 (2012).
(11) N.J. Thompson, S. Rosati, and A.J. Heck, Methods 65, 11–17 (2014).
(12) S. Rosati, Y. Yang, A. Barendregt, and A.J.R. Heck, Nat. Protoc. 9(4), 967–976, DOI: 10.1038/nprot.2014.057 (2014).
(13) A. Schmidt, U. Bahr, and M. Karas, Anal. Chem. 73, 6040–6046 (2001).
(14) N. Tahallah, M. Pinkse, C.S. Maier, and A.J. Heck, Rapid Commun. Mass Spectrom. 15, 596–601 (2001).
(15) I.V. Chernushevich and B.A. Thomson, Anal. Chem. 76, 1754–1760 (2004).
(16) F. Sobott, H. Hernandez, M.G. McCammon, M.A. Tito, and C.V. Robinson, Anal. Chem. 74, 1402–1407 (2002).
(17) R.H. van den Heuvel, E. van Duijn, H. Mazon, S.A. Synowsky, K. Lorenzen, C. Versluis, S.J. Brouns, D. Langridge, J. van der Oost, J. Hoyes, and A.J. Heck, Anal. Chem. 78, 7473–7483 (2006).
(18) Y. Yang, A. Barendregt, J.P. Kamerling, and A.J. Heck, Anal. Chem. 85, 12037–12045 (2013).
(19) A.D. Nisbet, R.H. Saundry, A.J. Moir, L.A. Fothergill, and J.E. Fothergill, Eur. J. Biochem. / FEBS J. 115, 335–345 (1981).
(20) J.A. Huntington and P.E. Stein, J. Chromatogr. B: Biomed. Sci. Appl. 756, 189–198 (2001).
(21) H. Nomoto, K. Yasukawa, and Y. Inoue, Biosci., Biotechnol., Biochem. 56, 1090–1095 (1992).
(22) D.J. Harvey, D.R. Wing, B. Kuster, and I.B. Wilson, J. Am. Soc. Mass Spectrom. 11, 564–571 (2000).
(23) S. Rosati, R.J. Rose, N.J. Thompson, E. van Duijn, E. Damoc, E. Denisov, A. Makarov, and A.J. Heck, Angew. Chem. 51, 12992–12996 (2012).
(24) S. Rosati, E.T. van den Bremer, J. Schuurman, P.W. Parren, J.P. Kamerling, and A.J. Heck, mAbs 5, 917–924 (2013).
(25) G.C. Gil, B. Iliff, R. Cerny, W.H. Velander, and K.E. Van Cott, Anal. Chem. 82, 661–6620 (2010).
(26) S. Tep, M. Hincapie, and W.S. Hancock, Carbohydr. Res. 347, 121–129 (2012).
(27) T.S. Raju and W.R. Strohl, Expert Opin. Biol. Ther. 13, 1347–1352 (2013).
(28) X.Z. Wang, V.W. Coljee, and J.A. Maynard, Curr. Opin. Chem. Eng. 2, 405–415 (2013).
(29) S.K. Rasmussen, H. Naested, C. Muller, A.B. Tolstrup, and T.P. Frandsen, Arch. Biochem. Biophys. 526, 139–145 (2012).
(30) S.K. Rasmussen, L.K. Rasmussen, D. Weilguny, and A.B. Tolstrup, Biotechnol. Lett. 29, 845–852 (2007).
(31) J. de Kruif, A. Kramer, R. Nijhuis, V. van der Zande, R. den Blanken, C. Clements, T. Visser, R. Keehnen, M. den Hartog, M. Throsby, and T. Logtenberg, Biotechnol. Bioeng. 106, 741–750 (2010).
(32) S. Rosati, N.J. Thompson, A. Barendregt, L.J. Hendriks, A.B. Bakker, J. de Kruif, M. Throsby, E. van Duijn, and A.J. Heck, Anal. Chem. 84, 7227–7232 (2012).
(33) N.J. Thompson, L.J. Hendriks, J. de Kruif, M. Throsby, and A.J. Heck, mAbs 6, 197–203 (2013).
(34) S.Z. Zhang, J.W. Xie, and C.S. Liu, Anal. Chem. 75, 91–97 (2003).
(35) H.J. Maple, R.A. Garlish, L. Rigau-Roca, J. Porter, I. Whitcombe, C.E. Prosser, J. Kennedy, A.J. Henry, R.J. Taylor, M.P. Crump, and J. Crosby, J. Med. Chem. 55, 837–851 (2012).
(36) R. Winkler, Rapid Commun. Mass Spectrom. 24, 285–294 (2010).
(37) Y.H. Tseng, C. Uetrecht, A.J. Heck, and W.P. Peng, Anal. Chem. 83, 1960–1968 (2011).












