ICP-MS: A Universally Sensitive GC Detection Method for Specialty and Electronic Gas Analysis
Application Notebook
The authors discuss the use of ICP-MS as an effective chromatographic detection method that is relatively easy to interface to gas chromatography for gas analysis.
Inductively coupled plasma–mass spectrometry has been investigated as a detector for gas chromatography and applied to analysis of trace impurities in specialty and electronic gases. Several key attributes, including high sensitivity, near-universal nature, ease of interfacing to gas chromatography, tolerance to matrix gases, and use of wet-plasma conditions to facilitate indirect calibration for a gas species for which a gas standard is unavailable, are discussed. Examples such as sulfur speciation in hydrocarbon gases at low part-per-billion levels demonstrate its use for specialty gas analysis. A more detailed case study is given on detection of trace impurities in arsine, including silane, carbonyl sulfide, hydrogen sulfide, germane, and hydrogen selenide. Under optimized conditions, detection of germane is possible down to 5 ppt.
As microelectronic devices become more complex and the features shrink, there has been increased demand for higher purity process gases. Some impurities, even if present at trace levels, can become incorporated into deposited layers and affect device performance significantly. Similar purity levels are required in other industrial specialty gas applications, such as for polymer manufacture, where trace impurities in monomer raw materials can affect catalyst activity significantly. This need to measure impurities at parts-per-billion and even parts-per-trillion levels has driven the development and use of highly sensitive and selective instrumental methods such as gas chromatography with inductively coupled plasma–mass spectrometry (GC–ICP-MS) and atomic emission detection (GC–AED).
ICP-MS originally was designed primarily to replace atomic absorption and ICP-optical emission instrumentation for analysis of metals in aqueous and organic matrices. However, its potential as a chromatographic detector was clear from the outset. Indeed, as the technique developed and matured, so too has the number of publications in which it has been used, as both a detector for liquid chromatography (LC) and GC (1–4). Like the AED, the ICP-MS can measure, with few exceptions, all the elements on the periodic chart. However, for most elements it typically offers greater sensitivity than the AED. The AED is the object of comparison because it is the only other commercially available GC detector that has similar specificity, selectivity, and sensitivity characteristics as ICP-MS (5,6). Table I compares the detection limits of the two technologies.



Table I: Comparison of detection limits for ICP-MS and AED
As a detection method, ICP-MS cannot provide a satisfactory replacement for the analysis of those elements that are in the atmosphere, such as N, O, C, and of course Ar, because it is an open system. Furthermore, it cannot measure fluorine at all due to fluorine's ionization potential. However, ICP-MS is comparable to other sulfur detection methods and about the same for chlorine, phosphorus, and silicon as AED. AED excels at elemental carbon measurement and is satisfactory for measuring fluorine. For most other elements, ICP-MS is just short of spectacular.
The excellent sensitivity of ICP-MS is complemented by the fact that it not only has high specificity over carbon-bearing matrices, but it is also very tolerant of other matrices. Venting is only required to avoid torch contamination as in the case of metallic deposition from metal hydride gases or simply to keep the torch pristine for subsequent work. It is relatively easy to interface a GC system with ICP-MS for volatile gas analytes, as shown in Figure 1. The heated transfer line becomes more important for less-volatile components, as transfer from the column to the plasma must occur at elevated temperatures to ensure that species remain volatilized in the gas phase until they reach the plasma.
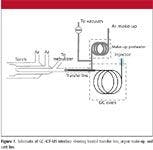
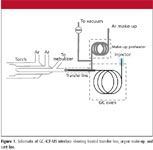
Figure 1
Experimental
The instrumentation used to generate the data used in this article consisted of a Thermo Electron X-Series ICP-MS system coupled with a Thermo Electron Focus gas chromatograph (both from Thermo Fisher Scientic, Waltham, Massachusetts). The gas samples were injected via a switching valve and a 400-μL sample loop into a 0.53-μm i.d. capillary GC column that was held isothermal during the analysis. The stationary phase was selected to optimize analyte separation. Column temperatures ranged from 35 °C to 70 °C, and the detector interface was set 10 °C above the column temperature. In addition, the column effluent was mixed with heated argon make-up gas to aid in transfer of the components into the ICP-MS system. A vacuum could also be applied at this point to remove undesirable components (such as the matrix peak) before entering the plasma (Figure 1). The ICP-MS system was operated using a nebulizer flow rate of 0.68 L/min, an auxiliary gas flow rate of 0.70 L/min, and cool gas flow rate of 13.5 L/min. Standard wet plasma tune conditions were employed to tune the ICP-MS system. Detection was achieved using single ions for P (m/z 31), Si (m/z 28), As (m/z 75), S (m/z 48), Ge (m/z 74) and Se (m/z 78).



Figure 2
Application
The chromatograms in Figures 2–4 illustrate a few of the routine measurements that can be made using GC–ICP-MS that demonstrate equivalent or better sensitivities than GC–AED. They also show the tolerance the plasma has for the matrix as well as its selectivity with respect to carbon. Sulfur speciation in hydrocarbon matrices can be accomplished at low parts-per-billion levels with very little interference. Figure 2 shows the minimal matrix contribution from a propylene sample. The only effect is a small baseline depression from the propane and propylene matrix. This also exemplifies use of collision cell technology. Under normal plasma operating conditions, there is a great deal of OO+ ion formation leading to interference with sulfur (OO+ and S+ both have the same mass of 32). A technique to reduce this interference is to add a small amount of oxygen in the collision cell after initial ion formation to form SO+ , thus forming mass 48 and eliminating the interfering OO+ (mass 32) from the indigenous oxygen. The collision cell approach varies and goes by different names according to each manufacturer but is somewhat analogous to chemical ionization used in organic mass spectrometers.

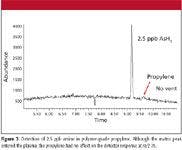
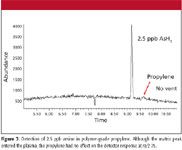
Figure 3
The lack of matrix interference and generally inherent high sensitivity of the ICP-MS system allows for extremely low detection limits in some applications, such as the analysis of arsine impurities in propylene as shown in Figure 3. Propylene exhibits virtually no signal, and subsequent sample analyses show no deterioration in response for arsine. This represents a detection limit for arsine approximately 10 times that which can be achieved with GC–AED. Figure 4 illustrates the worse case matrix interference when measuring phosphine in propane/propylene mixture. However, selectivity is still >100,000:1 P to C.

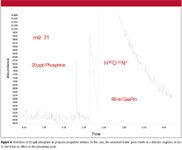
Figure 4

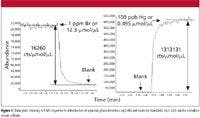
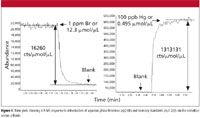
Figure 5
Cross Calibration
An ICP-MS system can be run in two modes when interfaced with a gas chromatograph — wet plasma or dry plasma. In dry plasma mode, the GC column effluent is connected directly to the torch and introduced with a make-up gas at a flow rate high enough to penetrate the plasma. Optimization of the ICP-MS system operating conditions can be accomplished using Xe (or other dopants) in the make-up gas. In wet plasma mode, a wet aerosol from the nebulization system is merged with the GC effluent before its entry into the plasma. Time and space do not allow for a detailed discussion of the advantages of both modes, but wet plasma has two advantages when analyzing low boiling simple matrices. First, it allows for a more robust plasma, since aspiration of water introduces an oxygen source and allows for operation at full power. The other advantage is that it allows us to indirectly calibrate for a gas species for which we might not have a gas standard. For example, if we wanted to determine the vapor phase concentration of mercury in the gas phase and only had a standard of a brominated compound, we would start by presenting known aqueous phase bromine and mercury standards to the instrument to determine the responses, as shown in Figure 5. The ICP-MS response to a 1-ppm or 12.3 μmol/μL Br solution was 200,000 counts, which converts to 16,260 counts/μmol/μL versus a blank. In the same way, the ICP-MS response to a 100 ppb or 0.495 μmol/μL Hg solution was 650,000 counts or 1,313,131 counts/μmol/μL versus a blank.

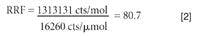
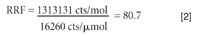
Performing the math in equation 1, we determine a relative response factor of 80.7.
The next step would be to analyze a known brominated hydrocarbon and the unknown mercury vapor, as demonstrated in Figure 6.
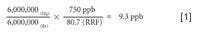
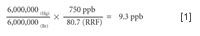
Completing the math in equation 2 we find that the unknown mercury is 9.3 ppb.



Figure 6
A thorough investigation of this strategy has not been made, but rough estimates have shown the accuracy to be within 50%. This normally would not be considered particularly good, but when dealing with concentrations for which standards are virtually unavailable at parts-per-billion and parts-per-trillion levels, it becomes very palatable.
Detection Sensitivity
In the mid-1990s we were presented with the problem of determining the concentration of germane impurity in bulk arsine. At that time detection limits were about 30 ppb using GC–MS and about 5–10 ppb using GC–AED or GC with reductive gas detection (RGD). This meant that detection limits were close but not quite good enough to make a measurement with confidence. So, a packed-column GC system was interfaced to an older ICP-MS system for the analysis. Coupling the two instruments together, it was possible to prove that the product contained 7 ppb germane. At the time, it was believed this hyphenated approach was a unique circumstance and that such a marriage of technologies was far too expensive to be used in any routine fashion. Times change, demands are greater, and specifications that were once 10 or 1 ppm are now parts-per-billion or parts-per-trillion levels. Many polymer-grade gas specifications are 10 ppb or lower for metal hydrides, and purities for highly pure electronic gases are even more demanding. This is beyond the reach of almost any other measurement strategy.
Impurities in Arsine: A Case Study
Arsine is an important gas used for metalorganic chemical vapor deposition (MOCVD) of GaAs and AlGaAs epitaxial films in the manufacture of high-speed microelectronic and optoelectronic devices. Since the purity of epitaxial films is critical to the performance of devices, microcontamination from O-, C-, S-, Si-, Se-, and Ge-containing species that cause unintentional doping, lattice defects, and oxide formation must be minimized. Unfortunately, critical contamination levels are not always above the detection limits of standard instrumentation. So, to ensure that these are avoided in the arsine process gas, development of sensitive analytical methods is required. Advances have been made to determine the carbon content indirectly by determining the amount of C3–C5 hydrocarbons. Flame ionization detection has been used historically to determine these but is limited to ~50 ppb without preconcentration. Application of pulsed discharge helium ionization detection has brought this detection limit down to a more acceptable range of 4–8 ppb. The hydride gas impurities SiH4, PH3, GeH4, and H2Se, and the sulfur compounds COS and H2S can all be determined at parts-per-billion and parts-per-trillion levels using GC–ICP-MS. Figures 7–12 demonstrate SiH4, PH3, COS, and H2S determinations.



Figure 7
The detection limits for silane, phosphine, carbonyl sulfide, and hydrogen sulfide are equivalent or only slightly better than what one could obtain using GC–AED as a comparison. It so happens that these are acceptable detection limits for these particular contaminants at this time. However, this is not the case for germane.



Figure 8
For certain devices, the critical contamination level of germane in arsine can be at the low single digit parts-per-billion level, which is just beyond the practical reach of GC–AED. Germanium is an n-type impurity of concern in GaAs and AlGaAs epi layers and is particularly detrimental to high-speed transistor devices such as high electron mobility transistors (HEMTs) and field-effect transistors (FETs). It has also been positively identified in GaAs layers using secondary ion MS (SIMS) analysis and its presence increases leakage currents and reduces the gain of transistors, which is undesirable. The common source of this contaminant is indigenous presence in the bulk arsine. Although GC–AED sensitivity for germane is generally acceptable, greater sensitivity is required in certain circumstances. Work on germane involved instrumental modifications designed to enhance sensitivity. In the course of this work, a linearity study was made on germane.


Figure 9
Figure 9 is a log–log plot of area counts vs. germane concentrations. Sample injections were made in triplicate at concentrations of 190 ppt, 1.9 ppb, 19 ppb, 190 ppb, and 1 ppm. Response was linear over five orders of magnitude. The initial effort using standard conditions resulted in a detection limit of 43 ppt based on 3 sigma in a population of seven. Figure 10 represents chromatograms of spiked and unspiked arsine. After instrument modification and retuning, a detection limit of ~5 ppt was established.



Figure 10
GC–ICP-MS analysis of hydrogen selenide has also been investigated. This analyte presented a different challenge and learning curve due mainly to its limited chemical stability. H2Se is one of the most labile metal hydrides. Thus, handling of standards and sample introduction are still proprietary. It also was a more difficult target since its elution order was after arsine instead of before as for the other hydrides. Once again, due to the forgiving nature of the ICP-MS plasma, it was possible to make this measurement. Figure 11 shows chromatograms of pure arsine with and without H2Se spikes.



Figure 11
Even under highly optimized conditions we were only able to achieve a detection limit of ~0.8 ppb H2Se. Investigation of an aqueous sample containing 200 ppb selenium and germanium (Figure 12) showed that Ge had about 10 times better response than Se. It is hypothesized that the higher detection limit is due to less efficient ionization, additional possible interference from krypton present in the argon, and 38 Ar40 Ar+ isotope background.

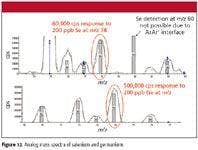
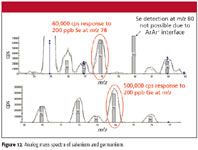
Figure 12
Conclusions
GC coupled with ICP-MS offers ultratrace sensitivity for many volatile metallic compounds, particularly those compounds of interest in semi-conductor manufacturing. In addition, ICP-MS is also technically competitive for detecting sulfur-containing compounds. Its element selectivity, cross-calibration capability, and ability to handle introduction of difficult matrix gases make it flexible and powerful as a chromatographic detection method and one that is relatively easy to interface to GC for gas analysis.
Since ICP-MS can be used in wet plasma mode, it can fill a role as a GC detection method while maintaining full ICP-MS analysis capability for aqueous samples.
From the point of view of development of ICP-MS as a GC detection method, areas that still need attention include reducing the overall complexity and dimensions of the instrumentation, improving software to truly integrate operation and data handling aspects, and investigating cross-calibration correlation techniques further.
William M. Geiger is with Consolidated Sciences, Ltd., Pasadena, Texas; bill@conscicorp.com. Mark W. Raynor is with Matheson Tri-Gas, Inc., Longmont, Colorado; mraynor@matheson-trigas.com.
References
(1) J.C.A. Wuilloud, R.G. Wuilloud, A.P. Vonderheide, and J.A. Caruso, Spectrochim. Acta, Part B 59, 755–792 (2004).
(2) D. Glindemann, G. Ilgen, R. Herrmann, and T. Gollan, J. Anal. At. Spectrom . 17, 1386–1389 (2002).
(3) B. Bouyssiere, P. Leonhard, D. Profrock, F. Baco, C.L. Garcia, and S. Wilbur, A. Prange, J. Anal. At. Spectrom . 19, 700–702 (2004).
(4) S. Nelms, ICP Mass Spectrometry Handbook (Blackwell Publishing Ltd., CRC Press, Oxford, UK, 2005).
(5) P.C. Uden, J. Chromatogr. A 703 (1–2), 393–416 (1995).
(6) S.R. Goode and C. Thomas, Spectroscopy 9(9), 14–22 (1994).


















Inside the Laboratory – The Petrochronology Group at University of California, Santa Barbara
January 29th 2024In this edition of “Inside the Laboratory,” John Cottle, PhD, a professor of geology at the University of California, Santa Barbara, and a member of Spectroscopy’s Editorial Advisory Board, discusses his group’s most recent work using “laser ablation split steam” analysis to measure elemental concentrations and isotopic ratios in rocks and minerals.



