It's All About Timing
In addition to expressing his longing for a Star Trek "tricoder," columnist Ken Busch shows that the core processes of ionization, ion dissociation, and mass determination in mass spectrometry are all completed in very short times compared to the overall length of the analysis.



In "Star Trek," that handy tricorder device provides exactly the analytical result needed to advance the story, and, amazingly, accomplishes this task in just a few seconds of script time. No lengthy sample preparation, no time seemingly spent in analysis, an instantaneous data interpretation, and an irrefutable conclusion. These characteristics, at some level, reflect the desiderata that underlie the development of miniaturized sensor technology in modern analytical chemistry. Modern mass spectrometry (MS) seems far removed from those desiderata, and from that fictional world of instantaneous analysis in the palm of your hand. Our instrumentation seems bulky and complex compared with the compact tricorder. We have developed portable mass spectrometers, dating as far back as 1978 (1,2), but they are targeted to fairly specific and limited applications, and you might not want to carry the batteries for a terrestrial model in your backpack. However, for outer-space applications, at a high cost, mass spectrometers can be made smaller while maintaining extraordinary capabilities. We may look to these devices as the topic of a future column. For now, in a discussion of time and MS, we focus on an Earth-bound laboratory scale. We note that each gas chromatography–mass spectrometry (GC–MS) or liquid chromatography–MS (LC–MS) run typically consumes minutes to tens of minutes. Add time needed for sample preparation, including collection, concentration, transfer, and disposal, as well as collection of data into a presentation format, and it's clear that we might spend time in the holodeck waiting for results with our present-day technology.
But MS itself really takes very little time. Figure 1 presents a slice of time from the very short to the very long (the vertical axis is logarithmic), and places MS time in perspective. The composition in the figure is analogous to that which appeared as a "Mass Perspective" in the February 2006 column (3). The axis crossing is at 1 s; the points to the lower left represent shorter times, and the points to the upper right are longer times. Rather arbitrarily, the length of a GC–MS or LC–MS run is set at 1000 s and is marked on the figure. The average human lifetime also is marked (your lifetime might vary). The solid line to the lower left is the time scale from about 1015 s (length of processes for formation of an ion in electron ionization) to 103 –101 s (time required for instrumental measurement of a single mass spectrum). We provided a laboratory scale of operation in the previous "Mass Perspective" figure. From a time perspective, the laboratory scale represents our ability to measure spectroscopic events at short time scales (say 1012 s) and the ability to integrate data over extended periods of time (extending to perhaps 106 s). Mass spectrometric time is a subset of the 18 order-of-magnitude laboratory scale, except perhaps for the shortest processes of ion formation.
The 1000 s allotted for GC–MS and LC–MS is needed for the separation process (we can consider this a form of sample preparation), and the need to (ideally) present one sample component at a time to the mass spectrometer for analysis. The development of analytical MS–MS provided some early insight into how the separations time barrier could be bypassed, and meaningful information preserved in a complex mixture analysis. More recently, ultrahigh mass accuracy MS, combined with more powerful data processing, is providing another pathway through which meaningful results from a complex information space can be extracted rapidly (4). We consider here MS itself, separate from external separations or sample preparation considerations. To apportion time in MS, we start logically at the point of ion formation and end (rather arbitrarily) at the point of ion detection. Figure 2 is an expanded perspective on mass spectrometric time, arrayed as a timeline, and serves in this discussion as the focal point for related concepts of ion formation and dissociation, and ion analysis by MS. Because the first steps of this timeline focus on ion formation, the unique processes that characterize ion formation in electron ionization, chemical ionization, electrospray ionization (ESI), or matrix-assisted laser desorption ionization (MALDI) will each occur in a different window of time. Here, we use electron ionization (EI) as our model. Descriptions of the timing of other ionization processes are held for a later presentation.
In EI, a gas-phase neutral sample molecule M interacts with an electron moving (usually) with 70 V of kinetic energy. As the ionizing electron interacts with the electrons of the molecule, it causes the release of an outer shell electron:

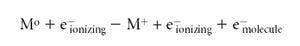
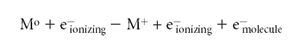
Note that the masses balance and the charges balance in this equation. Often, readers can find elementary equations that describe EI that are not balanced, and the fates of all the participants are unclear. The advantage of a proper equation is that it is made clear that there are two different types of electrons on the product side of the reaction, and that the energy of both can be measured to deduce the energetics of the ionizing reaction. The Franck-Condon principle (5) applies during the EI process. The removal of the molecular electron occurs in about 10-15 s (see Figure 1 as well), and is a "vertical" ionization process. Simply stated, this means that in this short period of time, the nuclei of the atoms that comprise the molecule (being much more massive than the electron participants) do not change in their positions. The molecule "suddenly" finds itself ionized, and retains its original structure. The ion often deals with an excess of internal energy by prompt dissociation. Because the structure is unchanged, these dissociations become a fingerprint of the original structure of the molecule. In this simple principle is the inherent usefulness of EI for structural studies by MS.

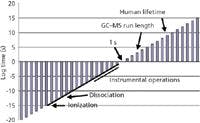
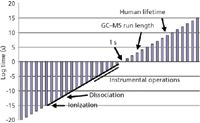
Figure 1: Overall time scale that shows ionic processes central to MS in perspective with the time required for typical GCâMS and LCâMS analyses.
Condon's scientific and professional contributions were many, and his prominence often targeted him out for special attention; see the review by Wang (6).
Before moving on to a more detailed discussion of timing for the processes of dissociation (and the reason why the "prompt" was added into the last paragraph), note that ionization by any of the other methods listed will take longer than does the process in EI. A proton transfer reaction that serves as the basis of chemical ionization requires the formation of a new chemical bond, and that will require about 10-12 s. The process in ESI requires that droplets be formed, that desolvation progress to the point of charge instability, and that the charge carriers ultimately associate, if not bind, with the originally neutral sample molecules. All of these processes take time, and the overall ESI process occurs within a window centered around 10-8 s, extending over several orders of magnitude. The width of the window reflects not only the complexity of a series of processes, but the population of molecules, each of which can follow a different path of different length. A similarly broad window applies in MALDI. Once the ions are formed, and are isolated in the vacuum of the mass spectrometer, however, subsequent processes of unimolecular dissociation may follow similar timelines.
How fast does dissociation occur? In answering this apparently simple question, we distill the results of extensive study in the 1950s and 1960s, which culminated in our understanding of bond-breaking processes in isolated, energized ions. The fastest that a specified bond can break is the time required for a single vibration of that bond, and this time is on the order of 10-12 s. Of course, most bonds that could break do not necessarily break on the first vibration, so there is again a time window over which prompt dissociations can occur. The breadth of that window is related to bond strengths, which is related to the nature of the bond and the bonding atoms. Our window of dissociation for simple cleavages is perhaps 10-12 to 10-10 s. In our elementary texts, we learn that not all fragment ions found in an EI mass spectrum result from simple cleavages. Rearrangement reactions that require the concerted breaking of one bond and the formation of a new bond also occur. The time window in which rearrangements occur is understandably somewhat longer, extending from perhaps 10-10 s to 10-8 s.
Most processes of ion formation and subsequent dissociation (through direct cleavages or through rearrangements) are all over in 10-8 s. The ions, once formed, are manipulated by electric fields to move into the mass analyzer of the mass spectrometer. There is a short physical distance to traverse (at least in quadrupole, sector, and time-of flight [TOF] instruments) from the source to the analyzer. The transit time must be brief, as must the subsequent passage of the ions through the mass analyzer, and the scanning of the mass analyzer. This restriction is placed on instrument operation by the fact that the mass spectrometer can be coupled to a separations method such as gas GC, and GC provides a stream of sample as eluting peaks that must be accurately sampled. For the total-ion current trace recorded by the mass spectrometer to be an accurate measurement of the eluents, the operation of the mass spectrometer must be complete in a very short time. In instrument-specific time values, the instrumental operational parameters are determinant factors. Consider a quadrupole-based mass spectrometer and typical voltages, physical sizes, and scan functions. The total time for passage of an individual ion from the source into the mass analyzer and then passage through the mass analyzer is on the order of a few tens of microseconds. This time is short compared with changes in sample concentration as peaks are eluted from a gas chromatograph, but orders of magnitude longer than the time required for ion formation and ion fragmentation. After EI in such an instrument, ions can be thought of as patiently waiting for something to happen. Remember that to record the complete mass spectrum, a quadrupole-based instrument will have to scan across a user-selected mass range. This operation usually takes a few tenths of seconds, still short enough to follow GC peaks as they are eluted from the column, but another eternity for the ions.
The situation is somewhat different for a TOF mass spectrometer. Simple math will show (again using the usual values for voltages and physical sizes) that ions traverse the flight tube within the 1–10 μs time frame (with the speedier, low-mass ions outracing the sluggish higher-mass ions). But because the mass analysis depends upon the fact that the race begins at the same time for all ions, the electronic pulse that starts the ions must be considerably shorter (on the order of nanoseconds). The importance of time-lag focusing (7) early in the development of TOF instrumentation, and insight into a number of new developments based upon timing that appeared in the 1990s, is immediately apparent. Rather than adding a scan function time on top of the ion transit time, the mass analysis is the transit time in a TOF instrument. There too, the time advantage becomes apparent through simple consideration of time perspective.

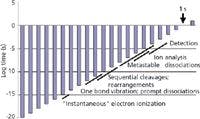
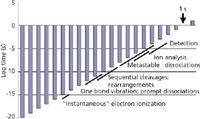
Figure 2: Timeline of ionic processes in MS from the point of ion formation to the point of ion detection.
There are more terms marked in Figure 2 and more concepts than can be considered here. We suggested that ions are formed very quickly in the vertical ionization process that is EI, and this is strictly true. We suggested that these ions dissociate (whether through cleavages or rearrangements) promptly, and that everything is over by about 10-8 s, and this is not strictly true. The rates of reactions for cleavages depend upon the amount of internal energy in the ion after its initial formation. Rearrangements and cleavages exhibit different internal energy dependencies. Energetic, hot ions (unstable ions) undergo prompt dissociations. Low-energy, cold ion (stable ions) do not dissociate. Ions of intermediate energy (metastable ions) are more leisurely in their reaction rates, and can dissociate when they are located physically in the mass analyzer itself. The timeframe for metastable ion dissociation overlaps that of ion analysis itself, as shown in Figure 2. In some analyzers, we can see the evidence of these postsource dissociation, and in other instruments, these ions are lost in passage. The study of metastable ions is a topic worthy of its own discussion. Other derivative topics that follow from a complete understanding of Figures 1 and 2 include field ionization kinetics, Rice-Ramsperger-Kassel-Marcus (RRKM) models, and the prospects for an MS-based tricorder. Stay tuned.
Kenneth L. Busch always wanted a tricorder when he was growing up (he still does, actually). Frustrated in this quest, he became interested in the field of metrology, and more recently in social metrology, defined as study of the expectations of the nonexpert about the acquisition and meaning of analytical results. Views expressed in this column are those of the author and not those of the National Science Foundation. He can be contacted at: wyverners@yahoo.com.
References
(1) R.W. Meier, Am. In. Hyg. Assoc.39, 233–239 (1978).
(2) C.M. Henry, Anal. Chem.71, 264A–268A (1999).
(3) K. Busch, Spectroscopy 21(2), 75–78 (2006).
(4) M.P. Barrow, J.V. Headley, K.M. Peru, and P.J. Derrick, J. Chromatogr., A 1058, 51–59 (2004).
(5) J.M. Standard and B.K. Clark, J. Chem. Educ. 76, 1363 (1999).
(6) J. Wang, ISIS 83, 238–269 (1992).
(7) W.C. Wiley and I.H. McLaren, Rev. Scient. Insrum. 26, 1150–1157 (1955).














Synthesizing Synthetic Oligonucleotides: An Interview with the CEO of Oligo Factory
February 6th 2024LCGC and Spectroscopy Editor Patrick Lavery spoke with Oligo Factory CEO Chris Boggess about the company’s recently attained compliance with Good Manufacturing Practice (GMP) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Expert Working Group (Q7) guidance and its distinction from Research Use Only (RUO) and International Organization for Standardization (ISO) 13485 designations.



