Periodic Reviews of Computerized Systems, Part I
Annex 11 to the EU's updated GMP regulations calls for periodic re-evaluation of computerized systems. This is what you need to know about the new rules.



In the first of a two part series, this month's column looks at interpreting the Annex 11 regulations and understanding the principles of a periodic review. The second part will discuss how to carry out the review and report it.
In my last Focus on Quality column we looked at the new European Union Good Manufacturing Practice (EU GMP) regulations, fousing on Annex 11 (computerized systems) and Chapter 4 (documentation) (1) that became effective on June 30 of this year. A new requirement in Annex 11 (2) is for periodic evaluation or periodic review. The regulation states in clause 11, "Computerized systems should be periodically evaluated to confirm that they remain in a valid state and are compliant with GMP. Such evaluations should include, where appropriate, the current range of functionality, deviation records, incidents, problems, upgrade history, performance, reliability, security, and validation status reports."
Some of the interpretations of this clause are
- You have to conduct periodic reviews of computerized systems.
- There are no exceptions to this regulation (for example, only critical systems need to be evaluated); all systems must be reviewed.
- The frequency of the review is determined by the company's quality assurance or laboratory management and justified where necessary to an inspector. Implicit in this statement is that you should apply a risk assessment to determine the frequency. Also, it is explicitly stated in clause 1 of Annex 11 that risk assessment is conducted throughout the life cycle of a computerized system.
- The aim of any review is to ensure that the system is compliant with the applicable regulations and that it remains in a validated state over the time period since the last review.
- The scope of the periodic review can cover the last full validation, change control records since the last validation, problems and their resolution, user account management, user training, and IT support such as data backup.
What is not stated in the regulation however, is the formality of the process. So how can we demonstrate to an inspector that periodic reviews have been carried out? Unless the reviews are formally documented, then you can't; it's as simple as that. So we will look at the overall process of a periodic review in this column and then in the next installment we will examine the practicalities of performing and reporting such a review.
A Small Regulatory Problem
Why perform a periodic review? This is probably one question that you may ask when reading the section above. In life, it is always better to use real examples to illustrate points you are trying to make because it demonstrates that other organizations can sometimes make bigger mistakes than you do. Take, for example, the following citation from a Food and Drug Administration (FDA) warning letter (3):
6. Your firm failed to check the accuracy of the input to and output from the computer or related systems of formulas or other records or data and establish the degree and frequency of input/output verifications [21 CFR § 211.68(b)].
For example, the performance qualification of your <redacted> system software failed to include verification of the expiration date calculations in the <redacted> system. In addition, there is no established degree and frequency of performing the verification. Discrepancy reports have documented that product labeling with incorrect expiration dates have been created and issued for use.
Your response states that you opened Investigation T-139 and you provide a January 29, 2010 through February 26, 2010 completion timeline. You have not provided a response to correct this violation and establish a corrective action plan to assure that computer systems are properly qualified.
The point is that the initial validation failed to check calculations in a computer system resulting in drug labels that were printed with incorrect expiry dates on them. Who picked up on the problem? The inspector! If the company had conducted a periodic review, this problem should have been identified by the reviewer, since it is an explicit requirement of the US GMP regulations, as noted in the warning letter. It is an obvious point for potential problems with any computerized system and the issue should have been identified and resolved long before the inspector strolled through the door for tea and cookies.
What's in a Name?
Although the Annex 11 regulation talks about a periodic evaluation (I have called it a periodic review), there are also laboratory audits carried out by the quality assurance departments. So are periodic evaluations, periodic reviews, or audits the same, or are they different? In my opinion, periodic reviews or periodic evaluations are one and the same, and are focussed on a computerized system, the process it automates, and the support processes for it. In contrast, an audit can cover a computerized system, a laboratory process, quality system, or a subset of any portion of the laboratory. Therefore, an audit can be the same as a periodic review or evaluation, but can also be a wider check of laboratory operations to ensure compliance with regulations and internal procedures. An audit also can cover computerized systems that are part of the process, but generally the scope of an audit is wider than a periodic review. Therefore, looked at from another perspective, periodic reviews are a subset of laboratory audits. Thus, in this column I will refer only to periodic reviews but this will include periodic evaluation and general audits that include computerized systems.
Overview of a Periodic Review
The topic of a periodic review that we will discuss is shown in Figure 1 and consists of two phases: planning and execution.
So we have established that the periodic review is an independent audit of a computerized system to determine if the system has maintained its validation status and, as said before, it is also a planned and formal activity. The first requirement for conducting a review is a standard operating procedure (SOP) covering the whole process. As a periodic review is a subset of an audit, the audit SOP should be relatively simple to adopt for a computerized system or you can use an existing audit SOP with a subsection for periodic reviews. The whole process should be described in the audit/review SOP and is shown in Figure 1.

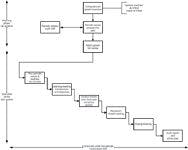
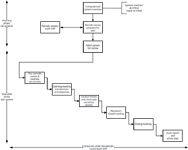
Figure 1: Flow chart for a periodic review or audit of a computerized system.
I have depicted the process as two parts:
- The planning phase (covering all systems). This links the inventory of computerized systems for the laboratory with a risk classification of each system (the figure suggests critical, major, or minor, but this is only a suggestion) to the SOP for periodic reviews and audits and the annual self-inspection schedule. The principle is that the critical systems are reviewed more frequently than major or minor systems, and major systems are reviewed more frequently than minor ones. The periodicity of review for each type of system is determined by your company.
- The execution phase (for each system reviewed). For each system selected for review there is a common process that consists of planning and preparation for the review, the opening meeting, performing the audit, including a check on the effectiveness of corrective actions from a previous review, the reviewer's closed meeting where the observations are examined to determine if there are any findings or noncompliances, the closing meeting where findings and observations are discussed, and finally the writing of the review report and an action plan for any corrective actions or preventative actions.
In this installment, we will focus mostly on the planning phase and will present the execution phase in an overview. The next installment will cover the execution phase in more detail.
Objectives of a Periodic Review
There should be two main goals for a periodic review of a computerized system:
- To provide independent assurance to the process owner and senior management that controls are in place around the system being reviewed and are functioning correctly. The system is validated and controls are working adequately to maintain the validation status.
- To identify those controls that are not working and to help the process owner and senior management improve them and thus eliminate the identified weaknesses. The impact of a finding may be applicable to a single computerized laboratory system or all systems in a laboratory.
It is the second objective that is the most important, in my view. Moreover, it is an important outcome from any periodic review for senior management to realize that some controls may require systematic resolution. If a problem is found in a procedure that is used for all systems, the resolution of this may affect all computerized systems used in the laboratory rather than just the one being reviewed.
Who Performs a Periodic Review?
Annex 11 does not say who should carry out the periodic review. So let's consider the possibilities:
- The process owner (the laboratory person who is responsible for the system)
- The system owner (the person responsible for the availability and support of the system)
- Quality assurance (QA)
Hmmm, I can guess some of your answers. The point I want to make is that people directly involved with a computerized system have a vested interest in their system and cannot make an objective decision if an activity is under control and in compliance or not. QA may be appropriate to conduct a periodic review, but the individuals have to know about computerized system validation and understand the regulations and company procedures in relation to computerized systems; not many in QA fit these criteria.
So to help us answer the question, what do the regulations say about this? European Union GMP chapter 9 (4) discusses self inspections (for example, audits and periodic reviews) in about two-thirds of a page, and the key elements of these regulations can be summarized as follows:
- Regulated activities should be examined at intervals to ensure conformance with regulations.
- Self inspections must be preplanned.
- Self inspections should be conducted by independent and competent persons.
- Self inspections should be recorded and contain all the observations made during the inspections.
- Where applicable, corrective actions should be proposed.
- Completion of the corrective actions should also be recorded.
So, from the perspective of the European regulations, we need a periodic review to be independent to ensure an objective and not subjective approach to evaluating your computerized spectrometer system. Indeed, the definition of independent is "not influenced or controlled by others; thinking or acting for oneself" (5). If a person who knew the system well were to perform a periodic review there is the possibility that he or she could miss something because it was familiar, that an independent reviewer could find. There is also the human tendency of a person involved in a system to focus on what they were doing well rather than the independent person who would be focusing on finding activities that were not compliant or could be done in a more efficient way. Therefore, independence of the person conducting a periodic review is of prime importance.
Skills and Training of the Reviewer
There are a number of requirements necessary for a person to effectively conduct a periodic review. These are
- Knowledge of the current good laboratory practice (GLP) or good manufacturing practice (GMP) regulations and the associated guidance documents issued by regulatory agencies or industry bodies. At a minimum, this knowledge needs to be in the same good practice discipline that the laboratory works to, but knowledge of the other requirements of good practice disciplines is an advantage because one discipline can be vague on a specific point that another might have more detail on. For example, in GMP there are no regulatory agency guidelines for the SOPs required for a computerized system, but the FDA has issued one for clinical investigations (6), where a minimum list of procedures can be found in Appendix A.
- Note that the knowledge of regulations above said "current." This criterion is included because the regulations and guidelines are changing at an increasing rate, especially in the European Union, where it is easier and quicker to change the regulations, as we have seen with Annex 11 itself (1).
- Experience working with computerized systems and knowing where (noncompliance) "bodies" can be buried and where bad practices can occur.
- An understanding of the current procedures used by the company, division, or laboratory for implementing and operating validated computerized systems. This is the interpretation of the regulations and guidance for the laboratory staff to work with that need to be both current and flexible.
- An open and flexible approach, coupled with the understanding that there are many ways of being in control. Therefore, the reviewer's ways of validating a computerized system and maintaining its validated state, or indeed those in the good automated manufacturing practice (GAMP) guide (7) and GAMP Good Practice Guide, Risk Based Approach to the Operation of GXP Computerized Systems (8) are not the only way of working. As Judge Jenkins stated in the case of the FDA versus Utah Medical, "Many roads lead to Rome"; the fact that a laboratory does something different to what you think it should do should not preclude it from being compliant (9). We will return to this point again when we discuss planning the audit.
- Finally, good interpersonal skills coupled with a hide as thick as an elephant's. The reviewer needs to ask open questions to understand what process is being carried out and investigate with pertinent questions to indentify if the work is adequate or if there are noncompliances. Persuasion may be required to change ways of working and a hide as thick as an elephant's may be needed to ignore any personal remarks or insults that may come your way.
So that outlines a periodic reviewer's skill set. Now the reviewers have to perform the review, which we will discuss in part II of this series.
How Critical Is Your System?
Putting the heading in a different way: Do we need to do a periodic review for all computerized systems? Well, the simplest answer to that question is to go back to clause 11 of Annex 11, quoted at the start of this column. It says "computerized systems," not critical ones or selected ones but all computerized systems. Therefore, this implies the need for categorization of computerized systems according to risk. Figure 1 shows the planning process, from the inventory to the annual schedule of periodic reviews to be conducted within an organization.
The starting point is the inventory of computerized systems contained in the laboratory validation master plan (10) that should be categorized according to risk. Some of the risk categories include critical, major, minor, no impact on good x practice (GxP), or high, medium, and low. You will want the most critical systems to be reviewed most often, as they pose the highest risk, and the lowest priority systems will be reviewed less frequently, as they pose the lowest risk. Most of the laboratory computerized systems featured in FDA warning letters are networked systems with multiple users, because they have the greatest impact.
From the inventory there will be developed a listing of the most critical systems: These will have the most frequent reviews to ensure that they are in control, with decreasing frequency for the major and minor systems. A review schedule for all computerized systems in the laboratory would be drawn up for the coming year. Typically, the schedule for the year will be written by the person responsible in QA the previous year and will list all systems to be reviewed and the months in which this will happen.
When to Perform a Review?
In my opinion, there are three or four possible times to review a computerized system:
- Before operational release of a system. This is to ensure that system development has been undertaken according to corporate standards and the validation plan. You may think that with all the planning and documentation produced in a validation, this is the last time to perform a review. However, I believe that this is the right time, for the simple reason that if something has been missed from the validation of a critical system, such as checking that a calculation works correctly, do you really want to operate a system for a year or so and then discover it? I thought not.
- Periodically, ensure that the operational system still remains validated. This will be a planned and scheduled activity and will be carried out on a regular basis for each computerized system in the laboratory while it is operational. There may be occasions when the review schedule is changed to link to a planned upgrade of the system, but this is typically performed after the system has gone live and is in operation.
- Before an inspection. Occasionally some companies may review a system before an inspection to ensure that there are no major compliance issues. However, if this approach is taken, ensure that the time between the audit and the inspection is sufficient to ensure that the remedial activities are implemented and work before the inspection. Personally, I believe that if a computerized system is truly critical it will be reviewed on a frequency that does not require a special review like this.
- When a system is retired and the data are migrated. I will not discuss this topic further in these two columns.
Although you can conduct a periodic review at these times during the lifetime of a spectrometer system, the principles of what a review consists of and the way one is conducted are the same.
Health Warning: Periodic Reviews Only Sample
Periodic reviews and audits should carry a health warning. It is important to realize that all reviews and audits are sampling exercises. The reviewer will select the procedures, documents, or records to examine and draw conclusions based on them that are applicable to the whole process being examined. Therefore, it is important to realize that noncompliances may exist where none have been found and reported, simply because the sample taken by the reviewer did not contain any problems. Trained reviewers and auditors know this and will inform the process owner of this, especially at the end of the review and also in the report. This is also known by GLP and GMP inspectors, and the FDA puts virtually the same text into all warning letters to deserving organizations:
The deviations detailed in this letter are not intended to be an all-inclusive statement of deviations that exist at your facility. You are responsible for investigating and determining the causes of the deviations identified above and for preventing their recurrence and the occurrence of other deviations.
There are two points to note: First, the specific statement indicating that the inspection is a sampling process and that the list of deviations from the regulations is never complete and cannot ever be unless the whole laboratory is reviewed. Second, and most important, is that the users, laboratory management, and quality assurance have the responsibility for ensuring regulatory compliance. If you find a problem, it is your job, and not that of QA or the inspectorate, to resolve it.
Therefore, if you want to hide behind a clean periodic review report, but know that noncompliant working practices are going on that the reviewer has not picked up on them, you are deceiving yourself. Moreover, it means that if an inspector identifies a problem, especially one that you have known of and done nothing about, it means that the subsequent corrective action will be more stringent than if you had found the problem yourself and fixed it under your terms. It is better for you to have found the problem and be in the process of fixing it, rather than for an inspector to find it, as it demonstrates that you are doing your job responsibly and diligently.
Slicing and Dicing: Defining the System Scope
It is important to get the scope of the audit correct and not to miss anything that could be significant in an inspection or could lead to questioning the quality of the results generated by the system (for example, unvalidated or incorrect calculations). Figure 2 shows an example of a computerized system used for quantitative bioanalysis in a GLP-regulated laboratory. The system consists of three high performance liquid chromatography (HPLC) systems with mass spectrometry (MS) detectors; each instrument has the MS software installed on a workstation to control the instrument and then acquire and interpret the chromatograms. Data are acquired directly to a central server that is supported by the IT department. In addition, there is a single workstation used by analysts to interpret chromatograms and relieve congestion on the instruments themselves for processing data.

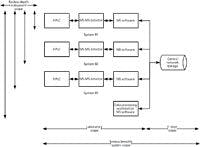
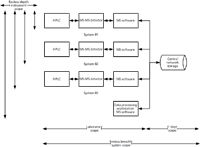
Figure 2: Scoping the periodic review of a computerized system.
The question is, How should a periodic review be scoped? From Figure 2 we can see that the system scope can be broken down into two parts. The breadth of the scope could include the portion of the system in the laboratory and the portion operated by the IT department. So the breadth of the audit needs to be decided: just the laboratory, just IT, or the whole system? As an aside, if the system had a server that is operated and maintained by the laboratory, then the whole system scope is the responsibility of the laboratory.
Then we need to determine the depth of the scope and determine how far to go reviewing the instrument and software aspects of the system. In the situation shown in Figure 2, each workstation has a separate installation of the MS software. Therefore, does the review take a sample from a single workstation and the attached instrumentation? From two, or all three installations? This is where risk management comes in. As the architecture of the overall system relies on three individual instances of the MS software that have to be set up (for example, users, access privileges, and software configuration) independently, do you want to know if the software instances are the same or not? How do you know that the three are the same? Although the installation and configuration documentation may say they are the same, has anything changed since then on one or more of the software instances? So, the depth of the review can depend on the technical aspects of the system — for example, individual installations of software with multiple software configurations versus a client server architecture where there is just a single configuration. Do not forget the data processing workstation as well, because it will be another individual installation and software configuration to consider.
Expressing a personal view, I would have a wide system breadth that would include both laboratory and IT aspects. The depth depends on the time available; in the case of Figure 2, I would review all instances to ensure equivalence, both on paper and in the software, but, again, this is dependent on the time available.
What is not shown in Figure 2 is whether the system has additional installations of the software for validation and training. When these are present, then the periodic review also needs to check them to see that they are correctly set up and equivalent to the operational system.
Also, consider an alternative to the system configuration in Figure 2: If the three systems were standalone with no reprocessing workstation, then the periodic review could cover three independent standalone systems. In this case, one aim of the review would be to demonstrate that the systems were equivalent and that similar results would be obtained from any one of them.

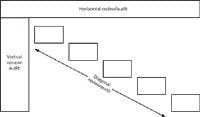
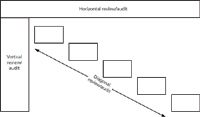
Figure 3: Types of audit or periodic review.
Types of Periodic Review
Ok, we now have the scope of the audit defined in terms of breadth and depth; what we now have to decide is how we will approach the review. There are three basic ways you could conduct a periodic review or audit, shown in Figure 3. These include the horizontal, vertical, and diagonal approaches:
- Horizontal audit or review: A horizontal review is conducted across the breadth of the system documentation; it attempts to cover all areas but at a low depth. It is typically undertaken if there is little time available to perform the review or if the system has not been reviewed before. The aim of this type of review is to give the confidence that all the major computer validation requisites are in place, but there may not be enough time to look in depth at how the various areas integrate together. Note that a horizontal audit can turn into a vertical audit if a problem is found during the review and the problem is required to be investigated in more detail.
- Vertical audit or review: In contrast to a horizontal audit, a vertical audit takes a very narrow perspective, selecting one or more areas to review and then going into a lot of detail, for instance checking that the controlling procedure has been followed. As an example, a vertical review could look at the change control procedure, and all the change requests for a specific system could be examined in much more detail than possible with a horizontal audit.
- Diagonal audit or review: As the name suggests, a diagonal review is a mixture of the horizontal and vertical audits. The purpose is to see that all the major validation elements are in place, operate correctly, and that all applicable processes work and are integrated together. Therefore, when examining the defining user requirements in the horizontal portion of a review, the diagonal audit will assess the traceability of requirements from the uniform reporting system (URS) to the rest of the life-cycle documents. In addition, the review can also take a test script and trace it back to the URS. As you can see, the diagonal audit examines the system more thoroughly than a horizontal audit.
In practice, all three types of auditing can be used effectively during a periodic review, depending on how much time there is available.
As mentioned above, a horizontal review can turn into a vertical one. For example, during a horizontal review of change control, the reviewer may ask to see three change requests selected at random from the list of change control requests; note the sampling process in the request. When examined and compared with the procedure, it may be found that two out of three requests did not follow the correct procedure. So the reviewer asks for three additional requests, again selected by the reviewer at random. When examined, two comply with the SOP and one does not. The reviewer now has a situation in which six change control requests have been reviewed and half comply with the procedure but, more worryingly, half do not. So what should the reviewer do? One alternative is to leave the change control process and complete the audit. The second is to dig further into the change control requests and the procedure to find out what is the true picture and leave the rest of the audit until the problem is investigated further. Because change control is such a vital mechanism for ensuring continued validation status, the archaeological excavation of the change control records should take precedence over the rest of the audit, in my opinion. Also, consider that there might be a systematic issue with change control that could affect all computerized systems in the laboratory. Therefore, the horizontal review turns into a vertical audit to discover how deep the problem goes: Has it been a consistent problem since the last review or has the problem only recently started?
Writing the Periodic Review Plan
Returning to the execution phase outlined in Figure 1, let's look at the various tasks in order, starting with writing the plan for the periodic review. This consists of a number of activities that cumulate in the plan:
- Agree on the date or dates for the review. When a system is due to be reviewed, the reviewer will contact the process owner to agree on dates for the review to take place. How long the review will take depends on the size and complexity of the computerized system, but typically it takes between 1–3 days. It is important that when dates are agreed on that key personnel will be available to discuss their specialist subject areas with the reviewer; otherwise the benefit of the review could be lost and noncompliances could be missed due to the lack of specialist knowledge. Smaller systems can be audited in a day but if the IT department needs to be included or a larger system is being audited, then it is more likely that 2–3 days will be required.
- Agree on the scope of the review. The reviewer should ask for information about the system (if not known already) to determine the scope of the review. This is particularly important for systems for which the laboratory and IT each have responsibilities. In such cases, the involvement of the two departments in the review needs to be coordinated. If the IT department is outsourced, then the outsourcing company needs to be contacted to ensure staff are available for the review.
- Write the periodic review plan. A review plan is written that contains the name of the system to be reviewed, agreed dates of the review, the department and location where the system is sited, and the regulations and procedures that the review will be based upon (for example GMP or GLP regulations and industry guidance documents). Included in the plan should be a timetable of activities of when the reviewer wants to discuss specific subjects. This is important to inform the laboratory and any other staff when their participation will be required. It helps them plan their own work for the day of the audit and avoids people hanging around waiting unneccessarily.
- Approval of the review plan. The periodic review plan is a formal document that needs to be signed by the reviewer as the author and a separate person as an approver. Depending on the company policies, the process owner may also need to sign the plan to acknowledge that he or she approves it and also to implement any corrective actions if there are any findings or noncompliances.
Preparation for a Periodic Review
What, I need to prepare for a periodic review? Yes! From the perspective of the reviewer, the individual needs to read up about the system and refresh his or her knowledge on any relevant SOPs that the system is validated and operates under. This means reading key documents such as
- computerized system validation SOPs
- validation plan for the last full validation of the system to be reviewed and the corresponding validation summary report
- user requirements specification for the current version of software that is installed
- organizational charts
- list of applicable procedures as a minimum and copies of some of the key SOPs, if possible, such as change control SOP and user SOPs.
This approach allows the reviewer to have an understanding of the system and procedures before arriving to perform the review and to be able to do some research, if required, before the review starts, thus saving time while on site. A reviewer could ask for more documents than listed above, but there is always a balance between the quantity of material and time used to prepare for the review and the time on site; personally I prefer to prepare using the key documents and procedures.
The spectroscopists who will be subject to the audit also need to prepare. At the most basic level, tidy up the laboratory (you will be surprised how many laboratories do not do this). Also, check that all documents are current and approved. If there are any unapproved or unofficial documents, they must be removed from desks and offices and destroyed. There are also other areas where the laboratory can prepare — for example, by reading the current procedures and ensuring that training records are up to date.
Activities During the Periodic Review
After the review plan has been written and approved and the reviewer has prepared for the review, the great day dawns and the review takes place. As can be seen in Figure 1, the activities that take place consist of the following steps:
- The opening meeting: This should be the shortest part of the periodic review, where the reviewer and the laboratory and, when appropriate, IT and QA staff who will be involved with the audit, are introduced to each other. The reviewer will outline the aims of the review along with a request for openness, as it is an internal audit designed to identify if the system is under control and remains validated or not. It is important that the head of the laboratory attend both the opening and closing meetings to see if there are any issues to resolve. The importance of the laboratory head attending must not be underestimated. If this key individual is not present, it sends a subliminal message to all, including the reviewer, that the individual has no interest in the validation status of the computerized systems. I certainly would document his or her absence in the review report.
- How do you work around here? I mentioned earlier that the person conducting a periodic review should not have fixed views of how a computer validation should be conducted, as there are many ways to be compliant. The key point in a periodic review is to keep asking the question, "Is the laboratory in control?" To orient myself for an audit or periodic review, after the opening meeting I prefer that the victims, sorry, auditees, give a short presentation of how validation is conducted in this laboratory. This approach is very useful as it allows the person conducting the periodic review (for example, me) to understand the terminology used by the laboratory and the overall validation strategy used for the system under review. In the long run, it helps to avoid misunderstandings and miscommunication between the two parties.
- Carrying out the periodic review. This is the heart of the periodic review where the last validation, organization, staff training records, change control, backup, and recovery operation of the system and associated procedures, and so forth will be assessed. We will look at this in more detail in the second part of this column.
- Reviewer's private meeting. This is the time for the person conducting the review to look over his or her notes and documents provided by the laboratory to see if any observations are identified as findings or noncompliances. Also, some outline notes for the closing meeting are prepared; these must include all major issues to be discussed with the process owner and laboratory management — the reviewer should not omit bad news and then add these little gems to the report so that it comes as a surprise to the laboratory.
- Closing meeting. This is where the reviewer will inform the laboratory staff about initial findings so that there will be no surprises when the draft report is issued for comment. All findings should be based on objective evidence, such as noncompliance with a procedure or regulation. There may be occasions when a difference comes down to interpretation of regulations. In that case, the auditor should seek information in industry guidance documents to support the finding.
- Write and approve the periodic review report and action plan. At the conclusion of the on-site portion of the review, the reviewer now has to draft the report, which will contain what the reviewer saw and the findings or noncompliances. Contained either in the report or as a separate document will be the action plan for fixing the noncompliances.
- Implementing corrective and preventative actions. Any findings and their associated corrective and or preventative action plans are implemented and monitored ready for review when the system comes due.
All the activities listed in this section will be discussed in more detail and the second part of a periodic review will be addressed in the next "Focus on Quality" column.
Summary
In this installment, I have looked at a periodic review or evaluation for computerized systems. The function is an independent audit to confirm that a system maintains its validated status and to identify any areas of noncompliance. In the next installment, I will discuss conducting the periodic review, who is involved, and reporting the observations and findings.
R.D. McDowall is principal of McDowall Consulting and director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com



R.D. McDowall
References
(1) R.D. McDowall, Spectroscopy 26(4), 24–33 (2011).
(2) EU GMP Annex 11, Computerized Systems.
(3) FDA Warning letter, AVEVA Drug Delivery Systems, Inc., 21 May 2010.
(4) EU GMP Chapter 9, Self Inspections.
(5) Webster's Dictionary (www.merriam-webster.com/dictionary).
(6) FDA Guidance for Industry, Computerized Systems in Clinical Investigations, 2007.
(7) GAMP Guide, version 5, 2008, Appendix 08, Periodic Reviews.
(8) GAMP Good Practice Guide: Risk Based Approach to Operation of GXP Computerized Systems, 2010, Section 12: Periodic Reviews.
(9) C. Burgess and R.D. McDowall, QA Journal 10, 79–85 (2006).
(10) R.D. McDowall, Spectroscopy 23(7), 26–29 (2008).















