Sample Preparation Problem Solving for Inductively Coupled Plasma-Mass Spectrometry with Liquid Introduction Systems: Solubility, Chelation, and Memory Effects
This tutorial on the chemical principles involved in dissolution, stability, and matrix components will help you successfully analyze a broad spectrum of metal analytes with ICP-MS using liquid introduction or flow injection.



In inductively coupled plasma–mass spectrometry (ICP-MS), problems persist in the determination of analytes that are commonly investigated as well as in specialty applications for those seldom considered by most analysts. Understanding the chemistry that is common to different groups of analytes permits the development of successful approaches to rinse-out and elimination of memory effects. It also equips analysts for development of successful elemental analytical approaches with a broad spectrum of matrices and other analytical challenges, whether the sample is solid or liquid.
Liquid introduction, in general, and flow injection, specifically, are the most widely used sample introduction methods for inductively coupled plasma–mass spectrometry (ICP-MS). Nevertheless, problems persist in the determination of analytes that are commonly investigated, as well as in specialty applications for those seldom considered by most analysts. Understanding the chemistry that is common to different groups of analytes allows the development of successful approaches to rinse-out and elimination of memory effects. This understanding also equips analysts for developing successful elemental analytical approaches in the face of a broad spectrum of matrices and other analytical challenges, whether the sample is solid or liquid.
The majority of ICP-MS applications for elemental analysis utilize liquid sample introduction whether or not the original sample was a liquid. The relative ease or difficulty of a given analysis depends on several factors, including the matrix and the chemistry of the analyte.
Probably the simplest matrix for elemental analytical purposes is water (that is, fresh water with low dissolved solids). Nevertheless, there are analysis problems with some analytes even in this matrix. General solubility rules state that alkali metal and ammonium ions are soluble in the presence of most anions. However, even in the presence of low concentrations of halide and polyatomic anions, many other metals hydrolyze and form poorly soluble hydroxides or oxides in water. When there is an absence of acid (or base in some cases), one may get an incomplete picture of the metal profile in a water sample because the poorly soluble hydroxide or oxide accumulates on walls of tubing, spray chambers, and nebulizers. An illustration of how such an incomplete profile may occur is presented in Figure 1. A total of 10 sequential water samples from an ultrapure water system were analyzed for 238 U intensity in counts per second (cps) in low resolution with a magnetic sector ICP-MS using a perfluoroalkoxy (PFA) 100 µL/min nebulizer and PFA double-pass spray chamber. If an analyst were to use uranium calibration standards diluted in ultrapure nitric acid before analyzing these samples, the uranium content would appear undetectable. A NIST 1643e water standard reference material (SRM) would give the appearance of validating the method accuracy. However, the NIST 1643 water SRM matrix contains 5% HNO3 to stabilize the characterized analytes in the water solution.

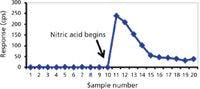
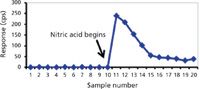
Figure 1: Illustration of the accumulation of poorly water-soluble metal oxides on sample introduction system surfaces from 10 ultrapure water samples versus continuous rinse-out when the water is acidified. The decline in 238U counts per second from sample 11 to 16 occurs as nitric acid dissolves and mobilizes the accumulated U from the surfaces.
Samples 11–20 in Figure 1 are acidified with 1% ultrapure nitric acid. It is obvious that the 238 U counts are elevated approximately 20-fold in all samples after the initiation of sample acidification. However, the approximate 100-fold increase in sample 11 demonstrates that 238 U from unacidified ultrapure water alone was accumulating in the introduction system. From that point on, the U continues to be mobilized and rinsed from the introduction system until it reaches a constant level. Thus, without proper sample preparation, sample carryover from accumulation could result in a false negative determination.
Figure 1 illustrates that the solvent and rinse solution for a given method must account for the analyte's aqueous chemistry. Nitric acid (1–5%) is commonly used for metal dissolution and stabilization for ICP-MS analysis. Nitric acid is a strong acid, and general solubility rules suggest that nitrates are soluble. The hydronium counter-anion from nitric acid is nitrate, thus it would superficially seem to be the universal solvent for metals. It is an appropriate choice for many inorganic analytes. Dilute nitric acid also is commonly used in diluents for urine analysis for this reason.
Memory Effects
There are metals, however, that are well known for memory effects when 1–5% nitric acid is used as a rinse solution. Memory effects are defined as the persistence of a given elemental signal after the analysis of a sample and a reasonable rinse time. A simple explanation for the causes of many memory effects can be made using the Pearson hard–soft acid–base (HSAB) theory as a model for coordination of a given metal (1). Though this dated model does not explain every case, it serves a useful purpose in general for approaching stabilization and rinse-out from an introduction system.
In the Pearson model, thorium is a very hard acid cation metal. Hard acid cation metals are preferentially coordinated by hard base anion ligands. If solid thorium oxide is placed in dilute nitric acid, it will slowly dissolve. The affinity of thorium for the oxygen ligands in neutral thorium oxide is strong. Because of this affinity, dissolution and stabilization of thorium as a hydrated cation in dilute nitric acid is not completely successful. Even thorium halides tend to precipitate as neutral hydrate halides over time. However, the addition of a dilute fluoride ion to a solution (when possible) may improve the rinse-out characteristics of thorium. Fluoride is a harder base anion ligand than oxygen and a better nucleophile than water oxygen atoms or other halides. For these reasons, fluoride can effectively compete with oxygen for coordination to thorium. However, even ThF4 is poorly soluble. When a sufficient concentration of fluoride is present for formation of anionic complexes such as [ThF6]2-, then the charged complex is quickly rinsed through the system without a memory effect. A rinse solution of 5% (v/v) nitric acid with 5% (v/v) hydrofluoric acid has been shown to eliminate thorium memory effects in urine at trace concentrations (2,3). Care must be taken because of the fluoride ion's neurotoxicity and the facile absorption via inhalation or dermal exposure. Safety concerns must be addressed when using sources of fluoride such as hydrofluoric acid or ammonium fluoride, and proper personal protection and appropriate fume hood ventilation must be used. In addition, the use of hydrofluoric acid necessitates an inert introduction system that includes a fluoropolymer nebulizer and spray chamber and a sapphire or alumina injector.
Mercury is an example of an element that exhibits memory effects for multiple reasons. According to Pearson's HSAB theory, mercury is a soft acid cation metal. Soft acid cation metals are more readily coordinated by soft base anion ligands, such as sulfur and halides other than fluoride. Soft acid cations also generally have higher electronegativities and more easily deformable orbitals, and they more readily form bonds with greater covalent character than hard acid cation metals. Therefore, mercury and similar metal ions form strong metal-sulfide and halide bonds. Environmental mercury commonly occurs in organomercury forms such as dimethylmercury and methylmercury halide. Even mercury(II) chloride has covalent characteristics in metal–chloride bonds. Because mercury(II) chloride is also linear and nonpolar, it is readily soluble in organic solvents such as ethyl acetate, pentyl acetate, and diethyl ether (4). This covalent, and sometimes nonpolar, character suggests an explanation for one cause of mercury's memory effect: adhesion to nonpolar surfaces such as polymeric tubing. Another cause of mercury's memory effect is that it is easily reduced. Nitric acid does not coordinate well with mercury and catalyzes its reduction in the presence of a reducing agent. Elemental mercury is neither lipophilic nor hydrophilic (5,6). Presumably, it migrates into crevices to escape either organic or aqueous solvents. Chelation of mercury ions with soft base anion ligands is a solution for avoiding such memory effects. Bromide and iodide ions strongly coordinate with mercury, but they form insoluble mercury halides. Chloride is more soluble and therefore is a better choice, and it aids in long term stabilization against reduction; but, as stated above, mercury(II) chloride is readily adsorbed on hydrophobic surfaces. Only when sufficient hydrochloric acid or another source of chloride is present at concentrations approaching 1 M or greater will mercury(II) be found predominantly in the [HgCl4]2- water-soluble anionic form. Such an excessive concentration of chloride ions would form Cl+ and ArCl+ species in the plasma and contribute to analyte signal suppression as a result of space-charge effects. In addition, such high concentrations of chloride may cause problems with rinse-out or stability of other elements such as thallium. Thus, though chloride from 1% HCl is sufficient to maintain long-term mercury stability in solutions, chelation with chloride may not be the most practical way to eliminate mercury's memory effect from a liquid introduction system.
One approach to eliminating the mercury memory effect is illustrated in a practical application with analysis of mercury in blood in alkaline solution with a chelating agent. The discussion of this application also addresses the analysis of metals in a more complex matrix than water or even urine.
Metals in Blood
Blood is a highly proteinaceous matrix containing hydrophobic lipids in cell membranes and in lipid transport proteins. Proteins generally do not tolerate acidic matrices well, specifically blood proteins. Acid causes precipitation and clumping of many blood proteins. Even slightly acidic solutions can have detrimental effects on blood consistency because the isoelectric point (pI) of many proteins, the pH at which the net protein charge is neutral, falls between pH 5 and 6. In this pH range, many proteins are as vulnerable to precipitation as in denaturing acidic solutions. In addition, most of the metals in blood are found in the cells or chelated by cell membrane functional groups. Therefore if the cells settle, or are not homogeneously dispersed, different analytical results could be obtained from the same blood sample. There are successful methods for elemental analysis of blood in acidic diluent, but additional precautions usually need to be made to ensure that nebulizers are not blocked and protein precipitate "strings" do not accumulate on or in the injectors.
A rugged method for elemental analysis of blood will take into account these aspects of the blood matrix, as well as the metals being analyzed. Freezing the sample until the day of analysis is beneficial because the freeze–thaw cycle ruptures many cell membranes. This allows for equilibration of cytosolic metals with the extracellular matrix, thus decreasing possible differences in sampling precision caused by clumps of cells. Before sampling blood, it should be thoroughly vortexed to disperse the cells. Detergents such as Triton X-100 (Dow Chemical Co.) are often used in blood metals analyses to solubilize and disperse lipid membranes in blood samples (7–9). Detergents also aid in membrane protein solubilization and dispersion. Although many proteins respond poorly to both concentrated base and acidic diluents, dilute base is generally better tolerated than dilute acid. For this reason, tetramethylammonium hydroxide has been used as an effective diluent for blood, usually in conjunction with Triton X-100 detergent (7–9). Unfortunately, many metal ions are not soluble in base. Therefore, a chelating agent is necessary when using a dilute basic diluent to minimize surface losses in the introduction system. For mercury and other soft acid cations, a chelating agent with sulfur ligands such as pyrrolidinecarbodithioic acid ammonium salt (APDC) provides excellent rinse-out while maintaining mercury(II) in the oxidized state (9). The use of a water-soluble compound with a sulfur ligand, thiourea, is shown in Figure 2 as an example of using a soft base anion chelator to rinse-out several micrograms per liter of mercury(II) spiked into blood from a PerkinElmer AS93 autosampler. It is compared to the rinse-out obtained with another commonly used combination (AuCl3 + EDTA). By eliminating the need for a high soft acid cation gold concentration (added in excess to compete with mercury for the reduction or chelation with ligands that form poorly soluble compounds), signal suppression because of space-charge effect in the ICP-MS is decreased, while accomplishing the same rinse characteristics as with the use of excess gold.

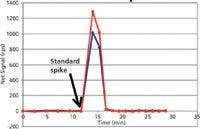
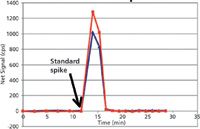
Figure 2: Hg washout: 0.01% thiourea, a soluble sulfur ligand (red), worked as effectively as 100 ppb gold(III) chloride + 0.01% EDTA (blue). The peristaltic pump was equipped with 0.45-µm i.d. PVC pump tubing. The liquid flow rate was 600 µL/min.
Chelating Ligands
Fluoride is not the best chelating ligand for all hard acid cations, and sulfur is not the only chelating ligand that can be used with soft acid cations. However, to offer a rough approximation, one can say that hard acid cations are chelated better with nitrogen, oxygen, or fluorine ligands, and soft acid cations are better chelated with sulfur or chlorine ligands. Figure 3 shows the hard and soft cation or anion characteristic of the most common oxidation states of many elements for analytical purposes (adapted from Wulfsberg [1]).



Figure 3: Cation acid and base anion characteristics for making appropriate choices of acid or chelating ligand in solutions and diluents. The charges shown in some cases are not a true representation of the form that would be found in solution. They represent only a common oxidation state.
Not all matrices are liquids. Laser ablation has come a long way as a quantitative technique for many solid samples in the last 10 years, many elemental applications are still better performed by somehow liquefying the sample. Depending on the sample, this may involve alkaline decomposition, acid digestion, or borate fusion.
Alkaline Decomposition
Alkaline decomposition is sometimes used to liquefy biological tissues. Carbohydrate, protein, or other biopolymers may be decomposed under alkaline conditions. This is not an oxidative digestion. Carbon is not eliminated as CO2, but solid biological samples may often be dissolved with an alkaline solution such as tetramethylammonium hydroxide as biopolymers are broken down by the base (10).
Alkaline decomposition rapidly decomposes even solid biological samples such as skin, nails, and hair (10). Unfortunately, it also degrades many organic chelating agents such as EDTA, DTPA, and APDC (11). Thus, when alkaline decomposition is used, analysts should be aware that organic chelating will need to be replaced after the decomposition or should be added after the dissolution of the matrix. It has been reported that free gadolinium (a hard acid cation) in tris hydrochloride buffer alone was lost during liquid chromatography–ICP-MS (LC–ICP-MS) analyses by accumulation in the size-exclusion column unless EDTA or DTPA (chelating agents with hard base anion oxygen ligands) was present. When hair was enzymatically dissolved near neutral pH, the chelating agents for gadolinium were not destroyed as they were during alkaline decomposition of hair (12).
Microwave Digestion
Microwave digestion is a common technique for bringing organic or inorganic solids into solution for analysis by ICP-MS. There are numerous variations on approaches to the acid mixtures used to accomplish oxidative digestions using a microwave system. There are too many to cite in a brief review, so only one good flexible model will be cited here. Environmental Protection Agency (EPA) Method 3052 is a very effective and flexible method for digestion of a wide variety of matrices. Typically, 100–250 mg of sample is digested at 180 °C for 9–20 min after a 5–10 min ramp. For simple matrices, the digestion is accomplished only with 9 mL of nitric acid. However, the option to add limited amounts of hydrogen peroxide, hydrochloric acid, and hydrofluoric acid depending on the matrix and the analyte requirements make it one of the most flexible methods available. One application, the digestion of smokeless tobacco samples, was accomplished using 9 mL of nitric acid, 0.5 mL of hydrogen peroxide, 0.5 mL of hydrochloric acid, and 0.5 mL of hydrofluoric acid (13). The additions were all within the limits of the method specifications. The principal acid, nitric acid, is an oxidizing acid for the destruction of organic matter, and one in which many metals will be very soluble as discussed earlier. However, it was noted that not all tobacco digestions were complete. Therefore, a small amount of hydrogen peroxide was added to support the complete oxidation of organic matter to CO2. Though colorless solutions indicated complete digestions, a white precipitate was noted; early work showed that lead and arsenic recoveries were low. The addition of 0.5 mL of hydrochloric acid produced a chloride ion to stabilize these analytes. The white precipitate was presumed to be insoluble silicates, because plants accumulate silicates for structural and other physiological purposes. The addition of 0.5 mL of hydrofluoric acid (see precautions described earlier) was sufficient to ensure that silicates were also dissolved in subsequent digestions. However, the addition of 0.75 mL or 1 mL of hydrofluoric acid caused another precipitation. Although calcium and magnesium are readily chelated by fluoride, their fluorides are not as soluble as those of some metals. Apparently, 0.5 mL of hydrofluoric acid was sufficient to dissolve the silicates, but an excess caused additional problems. This illustrates the importance of the optimum quantity of chelating ligand for effecting solubility, as described earlier for thorium. In the former case, the addition of too little HF would not sufficiently aid in the introduction system rinse-out, whereas in the latter case, too much caused precipitation of calcium and magnesium. These amounts had to be empirically determined.
Chelation Considerations
Before considering more challenging analyses and matrices, a fundamental understanding of the nucleophilicity of potential chelating ligands is often informative. In Table I, the chelation of several cations with EDTA is considered. As stated before, the charges on the cations are intended to represent the oxidation state and not the form in which they will be found in solution. Because the magnesium ion has a relatively low charge and large ionic radius relative to the other ions, its attraction to hydration-sphere water oxygen atoms is comparatively weaker than the attractions to oxygen in the other cases. Even though neither the carboxyl oxygens nor the tertiary amino groups of EDTA are strong nucleophiles, EDTA is readily able to compete with hydration-sphere water molecules for chelation of magnesium. Because of its relatively greater charge and smaller ionic radius, chromium(III) has a much greater attraction to oxygen atoms from water. The displacement of these ligands by EDTA or even by other water molecules has a half life of hours unless heated to increase the rate of coordination by EDTA. Titanium(IV) and silicon, however, are poorly chelated by EDTA. The binding of oxygen atoms by these oxides is very strong.

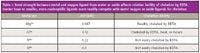
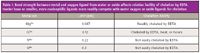
Table I: Bond strength between metal and oxygen ligand from water or oxide affects relative facility of chelation by EDTA. Harder base or smaller, more nucleophilic ligands more readily compete with water oxygen or oxide ligands for chelation.
Titanium(IV) oxide and silicon in silicates can be viewed together as examples of extremely hard acid cations — high oxidation states and very small ionic radii. Together, these characteristics make them challenging targets for dissolution whether they are considered analytes or as difficult matrix components. Because of their high oxidation states and effective nuclear charges, they have a very strong attraction to hard base anion ligands. In the compounds mentioned, the ligands are the oxygens in their three-dimensional structural forms. Water oxygens are not sufficiently nucleophilic to successfully attack the respective electrophiles and break the metal–oxygen bonds. Fluoride, usually from hydrofluoric acid (see precautions described earlier) is the most commonly used nucleophile for attacking the titanium(IV) or silicon ion because it is more strongly chelated as a hard base anion ligand for each cation in a water matrix than the water oxygens. Water would only be hydrolyzed by these hard acid cations anyway. An example of the use of hydrofluoric acid to dissolve silicates was described above in the analysis of tobacco.
Hydroxide, the conjugate base of water, is also much more strongly nucleophilic than water. Basic conditions have been used to dissolve silicates because hydroxide may successfully attack the silicon-oxygen bonds. Thus, the analyst who cannot use hydrofluoric acid for instrumental or safety reasons has an alternative. However, the analyst must consider the solubility of many cations in base, and the fact that organic chelating agents are labile to concentrated hydroxide.
Borate Fusions
An example of an application in which silica and other oxides were analyzed in a titanium(IV) oxide excipient matrix used an approach to aluminum oxide and silica dissolution with basic borate fusion (14). This approach includes heating with potassium hydroxide and borate from boric acid. As discussed, hydroxide is a strong nucleophile, but heating weakens the metal–oxygen bonds and increases the nucleophilic reaction rate. Even though the fusion breaks down the titanium, aluminum, and silicon oxide lattices, the addition of water permits some reformation of insoluble oxides after hydrolysis. Borate, however, is able to disperse between the various oxides at the elevated temperatures and act as an intervening ligand to prevent reformation of a tight lattice, rendering the fused metals more soluble. Many borates also are very soluble. For the analysis of other metals from the matrix, one would still need to dilute and add an appropriate chelating agent. However, this example illustrates the use of appropriate chemistry to effect a solution to a matrix problem.
Conclusion
The most commonly used techniques for sample analysis with ICP-MS continue to be liquid introduction and flow injection. To utilize these standard techniques, analysts must render the matrix soluble and the analytes mobile in solution whether the sample was originally a solid or a liquid.
To successfully develop and perform analyses of a broad spectrum of metal analytes in solution from a variety of matrices, a general understanding of the chemical principles involved for dissolution, stability, and rinse-out for matrix components and analytes is helpful. However, it is difficult to know the chemistry of every element and every oxidation state in the periodic table. Thus, adaptation of a general model to understand approaches to applications involving the dissolution, stabilization, and rinse-out of matrix components and analytes will aid in devising approaches for method development. The Pearson hard–soft acid–base principle was used as a general model to approach such problems in this review.
In general, this approach relies on the use of soluble hard base anion ligands for coordination of hard acid cation metals, and soft base anion ligands for coordination of soft acid cation metals.
Dissolution of matrix components may be accomplished utilizing techniques such as alkaline decomposition, microwave acid digestion, and borate fusions.
Once analytes are dissolved and appropriately stabilized in solution, the most challenging part of method development for liquid introduction systems is accomplished.
Acknowledgment
This tutorial was adapted from the first half of a course presented at the 7th International Conference on Sector Field Inductively Coupled Plasma Mass Spectrometry in 2008 and the 2012 Winter Conference on Plasma Spectrochemistry on sample preparation for liquid introduction systems.
The findings and conclusions in this report are those of the author and do not necessarily represent the views of the Centers for Disease Control and Prevention.
R. Steven Pappas is a research chemist and tobacco inorganics team lead in the volatile organics and tobacco branch, in the Division of Laboratory Sciences, at the National Center for Environmental Health, U.S. Centers for Disease Control and Prevention, in Atlanta, Georgia. Please direct correspondence to: RPappas@cdc.gov.



R. Steven Pappas, PhD
References
(1) G. Wulfsberg, Inorganic Chemistry (University Science Books, Sausalito, California, 2000), pp. 191–228.
(2) B.G. Ting, D.C. Paschal, J.M. Jarrett, J.L. Pirkle, R.J. Jackson, E.J. Sampson, D.T. Miller, and S. Caudill, Enviro. Res. 81, 45–51 (1999).
(3) R.S. Pappas, B.G. Ting, J.M. Jarrett, D.C. Paschal, S.P. Caudill, and D.T. Miller, J. Anal. Atomic Spectrom. 17, 131–134 (2002).
(4) S.N. Tandon and C.B. Gupta, Talanta18, 109–112 (1971).
(5) S. Okouchi and S. Sokichi, Bulletin of the Chemical Society of Japan 54, 2513–2514 (1981).
(6) Y.V. Alekhin, N.R. Zagrtdenov, R.V. Mukhamadiyarova, and A.S. Smirnova, Bulletin of the Department of Earth Sciences, Russian Academy of Sciences, doi. 10.2205/2011NZ000136 http://onznews.wdcb.ru/publications/v03/asempg11ru/2011NZ000136R.pdf (2011).
(7) J.A. Moreton and H.T. Delves, J. Anal. Atomic Spectrom. 13, 659–665 (1998).
(8) T. Delves, VAM Bulletin20, 16–21 (1999).
(9) W.J. McShane, R.S. Pappas, V. Wilson-McElprang, and D. Paschal, Spectrochimica Acta Part B 63, 638–644 (2008).
(10) I. Rodushkin and M.D. Axelsson, Sci. of the Total Environ. 250, 83–100 (2000).
(11) V. Loreti and J. Bettmer, Anal. Bioanal. Chem. 379, 1050–1054 (2004).
(12) R.S. Pappas, E.C. Obot, J.D. Thomas, and R.Y. Wang, "Gadolinium and Gadolinium-DTPA Complex Binding Studies in Hair and Plasma Proteins," presented at the 2010 Winter Conference on Plasma Spectrochemistry, Fort Myers, Florida, 2010.
(13) R.S. Pappas, S.B. Stanfill, C.H. Watson, and D.L. Ashley, J. Anal. Toxicol. 32, 281–291 (2008).
(14) M. Mutsuga, K. Sato, Y. Hirahara, and Y. Kawamura, Food Add. and Contamin. Part A. 28, 423–427 (2011).













