Units of Measure in Spectroscopy, Part III: Summary of Our Findings
Spectroscopy
What is it that we thought we knew that we have learned "ain't so" from the work reported in this series of columns?Volume 30 Number 2Pages 24-33What is it that we thought we knew that we have learned "ain't so" from the work reported in this series of columns?
Volume 30 Number 2
Pages 24-33
What is it that we thought we knew that we have learned "ain't so" from the work reported in this series of columns?
This column is the next installment in our discussion of units of measure, an outgrowth of our discussion of the classical least squares (CLS) approach to calibration (1–13). As we usually do, when we continue the discussion of a topic through more than one column, we continue the numbering of equations, figures, and tables from where we left off.
The quote "It isn't what we don't know that gives us trouble, it's what we know that ain't so," is variously attributed to Mark Twain (14,15) or Will Rogers (16,17). The question this quote brings to mind for us is: What is it that we thought we knew that we have learned "ain't so" from the work reported in this series of columns? We have discussed some of the answers to this question in previous columns, but others are described here.
Classical Least Squares Versus Beer's Law
The first thing we thought we knew was that CLS and Beer's law are the same. This equivalence is stated (mainly by assumption) in many places in the literature. While the CLS algorithm requires that Beer's law holds for the chemical system under consideration, they are quite different concepts. The CLS algorithm only relates spectra to spectra, and tells us that the spectrum of a mixture is a sum of the spectra of the components; there is no consideration given as to how those spectra arise.
Beer's law, on the other hand, tells us that the spectrum of a given chemical component in the mixture results from its intrinsic absorptivity at given wavelengths, and each contributes to the spectrum of the mixture in proportion to the concentration of the corresponding component. That Beer's law and the CLS algorithm are not the same is also easily seen when considering the units used in the two concepts. When discussing our results with various people, there would inevitably be a discussion about the units used in Beer's law, and the fact that the absorption coefficient (a) used in Beer's law is assigned units that cancel the units of concentration used. This way, the net units from the Beer's law expression A = abc cancel out, which they must since the absorbance (A) is dimensionless. Standard texts about spectroscopic analysis, such as Spectrochemical Analysis (18), specify the absorption coefficient in terms of the concentration. Indeed, fairly early on (page 20) in the specified book comes the statement, "If b is expressed in cm, a has units of (conc)-1 cm-1." To further emphasize the point, the absorption coefficient can have any of various numerical values, depending on the units used to express the pathlength and the analyte concentration.
That is all true, but immaterial. Let us compare the equations for the two concepts:

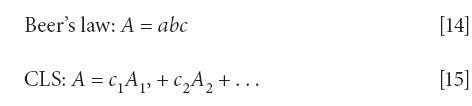
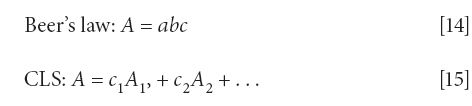
There is confusion, however, created by the use of the symbol "c" in both the Beer's law equation and the CLS equation. Since "c" is a multiplier of a in the Beer's law expression, and is also a multiplier of A in the CLS expression, the (natural, I suppose) assumption is that the two usages stand for the same quantity, especially since the two equations are so closely related. Furthermore, explanations of Beer's law often include a statement that the total absorbance (A) at any wavelength, is equal to the sum of the contributions from all components of the sample at that wavelength. We know that it is said, because before we knew better we said it ourselves on occasion, but the statement about the ". . . sum of the contributions . . ." is, strictly speaking, not a statement about Beer's law, but a statement about CLS.
The differences between the two equations are subtle, but important. First of all, Beer's law (equation 14) has only a single term on its right-hand-side, while the expression for CLS has multiple terms on the right-hand side. More importantly, as we described above, the concentration value for Beer's law (equation 14) can have any number of useful units. Of necessity, however, the coefficients of equation 15 must be dimensionless, since they are multipliers of dimensionless quantities (the absorbances), and the products of which must also be dimensionless (because they are also absorbances).
Also significant, the expression for Beer's law has a, the absorption coefficient, on its right-hand side, while the expression for CLS has A, the absorbance on the right-hand side. Since the variable labeled "c" is multiplied by different quantities, but the product of both expressions is absorbance, then it is clear that "c" cannot stand for the same quantity in both equations. Let us look at the differences a little more closely.
In equation 14 (Beer's law), c is multiplied by b and a, where c is defined as concentration. As noted above, the units for that concentration are not specified by Beer's law; the only requirement is that the units of concentration and the units for the absorption coefficient should cancel. This is more of a requirement on the absorption coefficient than on the concentration, however. Typically, in practice, units of chemical interest (weight %, molality, molarity, and so on) are used to express the concentration. In this case, the units for a, the absorption coefficient, must be such as to cancel the units used for concentration - for example, L·g-1cm-1 when the pathlength is expressed in centimeters and the concentration in grams per liter (18).
In equation 15 (CLS), however, the variable c is not multiplied by physical quantities as in equation 14, it is multiplied by A, which is also a (dimensionless) absorbance. Because that absorbance is already dimensionless, it is clear that c is also a dimensionless quantity. In equation 15, therefore, c is not a concentration, it is merely a coefficient that multiplies absorbances to express their contribution to the total absorption.
Both coefficient and concentration can plausibly use the same symbol, c, to express them, thus the confusion. But then what does the coefficient stand for, besides the spectral contribution? By trial-and-error experimentation, we found that if the concentration of the components in a mixture are expressed as volume percentages (or volume fractions), then the numeric values for the concentrations (also expressed as volume fractions) are numerically equal to the spectral coefficients that we determined for that mixture using the CLS algorithm.
This provides the justification for equating the CLS coefficients with the volume percentage concentrations, and to use those coefficients as surrogates for the concentrations expressed as volume percents, even though they are not the same things.
The Meaning of "Concentration"
The second thing we thought we knew (but that ain't so) was that "concentration" was a fixed, immutable property and that as a consequence, different expressions of it (that is, casting it in different units) are essentially equivalent except possibly for a scaling factor. We have quite belabored the point that this is not so in previous columns (10,12), showing that not only are the relationships between concentrations expressed in different units that are not linearly related, but also that there is not even a one-to-one correspondence between them. This dichotomy depends only on various extraneous physical or chemical properties of the mixture components and not on the spectroscopy. We have already pointed out that because of this relationship nothing you do to the spectra will correct the problem, all you can do is make the model you create fit the particular, unique set of nonlinear relations inherent in the sample set used for the calibration. We will not belabor the point further here.
What we will discuss, however, is an extension of this concept, to a broader range of utility. We can express the volume percent as follows:

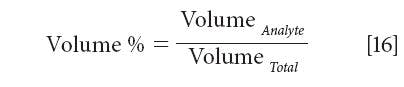
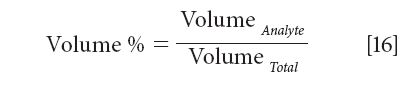
An important characteristic of the volume % that makes it useful for calibration with other algorithms is the fact that, by virtue of being equal to the spectroscopic measurement, it is ipso facto linearly related to the spectroscopic measurement. When using other algorithms, the critical characteristic needed is for the concentration units used to also be linearly related to the spectroscopic measurement. Multiplying volume percent by a constant, for example, might provide such a unit, which would therefore be satisfactory for calibration.
As an example, if the density of the analyte were known we could multiply the numerator of equation 16 by that value:

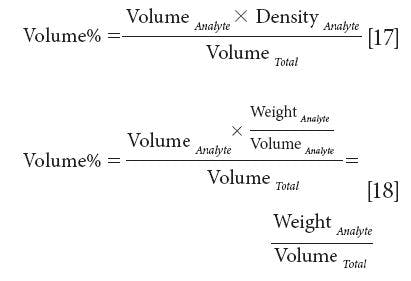
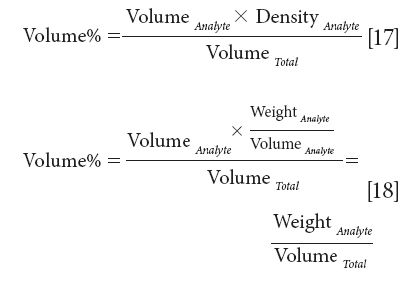
Thus, weight of analyte per unit volume of sample would be a suitable unit for expressing concentrations and that would provide a unit that was linearly related to the spectroscopy.
We could go even further. Consider dividing equation 16 by the molecular weight of the analyte (equivalent to multiplying by the inverse of the molecular weight, which is what we shall do):

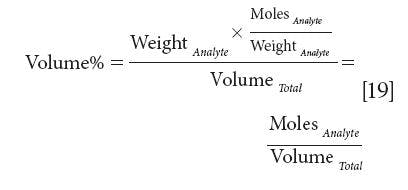
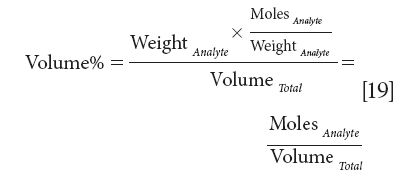
Thus, we wind up with a concentration expressed in units of moles per unit volume, which is, of course, the very chemically oriented unit we call molarity, and is thus a concentration unit that is linear with the spectroscopic measurement. We thank Jim Brown for pointing out these equivalencies to us.
It took the use of the CLS algorithm to determine that the spectroscopic behavior mirrored the volume percentages of mixture compositions, but clearly the physical processes involved are not dependent on the nature of the data processing algorithms applied to the data. Thus, while those algorithms that are considered the "standard" calibration algorithms (that is, partial least squares [PLS], principal components regression [PCR], and even multiple linear regression [MLR]) are commonly used, they are also affected by these physical effects.
Calibration Transfer
A third thing we thought we knew was that the difficulty of calibration transfer is because of physical interinstrument differences, and several recent columns have inspected that source of problems in detail (12,13,19–22). In fact, the problems encountered when trying to transfer calibrations from one instrument to another are not always solely because of the differences between instruments, but rather are because of the difference between sample sets. As described previously, the calibration process needs to approximate the nonlinear relations between the spectral measurements and the weight percents so often used to express the analyte concentrations. The corrections needed are unique to each sample set, and therefore are incorporated into the calibration model developed on that data set.
In an earlier column (23), we demonstrated that two instruments using the same calibration could provide the same predicted values for the same samples even when the constituent values they were calibrated against were random numbers. In other words, near infrared (NIR) values from a given set of samples agreed with other NIR values from that same set of samples even when the NIR values agreed with nothing else. But why would that be so? At the time, we had no explanation. Since we didn't understand what was happening "under the hood" so to speak, the only lesson we came away with from that exercise was to conclude that "NIR predicts NIR" and that, therefore, you should not use NIR analysis to provide reference values for developing a calibration, because the instruments could agree regardless of whether or not the calibration was any good.
In hindsight, and in the light of the current experiments, however, the answer is clear: both instruments were measuring the same samples and, more importantly, the same sample set. Thus, in this case the nonlinearities that affected the readings on either instrument were the same for both instruments, and the calibration modeled those nonlinearities very well. Therefore, the calibration calculated from one instrument is optimized for that set of nonlinearities and would predict those samples to have the same values on any instrument regardless of what those values represented, as long as the different instruments provided the same readings for each sample. This, after all, is indeed the meaning of "calibration transfer": to demonstrate that different instruments provide the same results for a set of samples. The accuracy, precision, and other test characteristics of the measurements are different issues, and they need to be addressed separately.
Previously, it was thought that the important differences were the ones between instruments, and the calibration transfer "problem" was attributed to the instruments. However, we now know that when the instruments are similar (that is, at least of the same type from the same manufacturer), they will respond similarly to the sample nonlinearities; so, it is the sample differences because of the use of different sample sets that are an important cause of the difficulties when incorrect units are used to express the analyte concentration, since each new sample set brings its own unique set of nonlinearities along with it. Therefore, the calibration for one instrument will work almost identically on another instrument of the same type, as long as it is measuring the same sample set.
Typically, calibration transfer exercises are invariably conducted using a different sample set than was used for calibrating the initial "parent" instrument, and this difference is still an important factor in verifying the ability of a calibration model to accurately predict new samples. However, this new sample set will bring along with it a new and different set of nonlinearities that would need to be compensated for, but the calibration model will not have those compensations built into it since they were not present in the original calibration sample set. This finding from our work indicates that it is the differences in sample sets, combined with the use of an incorrect measurement unit that, in a large measure, cause the difficulties in transferability. That being said, wavelength alignment, lineshape, linearity, and resolution differences must be small for calibration transfer to be successful, otherwise the instruments will not agree with each other even with the same samples.
Can anything be done about this? For liquid samples, at least, the answer is obvious: use volume percent as the measurement unit. Since that is what is directly related to the spectroscopic measurement, the effects of the nonlinearities disappear. For solids, and especially for powdered solids, the answer is not yet clear, although it would seem that a measurement unit more similar to volume percent than the current, almost universally used units, would minimize the nonlinear effects. Again in hindsight, we realize that this is how NIR data should behave; the same readings should give the same results, good or bad.
Some caveats here, though: First, the conclusions we reached only apply when Beer's law holds. Second, this is not to say that when differences do exist between instruments, those differences won't also prevent the transfer of a calibration. Some of those differences include differences in wavelengths between instruments, in detector linearity characteristics, and in the nature of the data (for example, Fourier transform-infrared [FT-IR] spectroscopy provides spectra linear in and having constant resolution in wavenumbers, but diffraction-grating-based instruments provide spectra linear in and having constant resolution in wavelength). Of course, other types of differences can also be found, but we won't go into those here. Any or all of these can prevent successful calibration transfer. Calibration transfer depends on a chain of events all occurring properly; a defect anywhere along the chain becomes the "weak link" that negates the rest of the chain. The only difference in the finding of the nonlinear relations between different units is that this is a new and previously unrecognized source of error, despite the fact that it can easily constitute the largest error contribution to a spectroscopic measurement.
Calibration transfer, however, is a problem that is separate and distinct (and perhaps easier) from calibrating and validating a calibration in the first place. To demonstrate calibration transfer, it is necessary to show that the same samples give the same results when measured on the different instruments. Since the use of a different sample set brings a whole new set of nonlinearities along with it, that only complicates the situation by confounding the differences between sample sets and the differences between instruments. To be sure, validating a calibration using a set of samples separate from the calibration set is still a necessary step in developing a calibration, but it should not be combined with the already-difficult procedure of demonstrating that different instruments produce the same readings.
Other Things We Thought We Knew
There are a number of other "facts" we thought we knew that this work indicates may or may not be facts at all, although the returns are not all in yet. Most of them have to do with the use of spectroscopic analysis in conjunction with chemometric methods of data analysis and quantitative calibration. Let us take an overview of these facts, as we know them (we think).
Extrapolation
It is common knowledge that quantitative spectroscopic calibrations cannot be extrapolated. We now know that this is caused by the nonlinear relationships between the volume percentage of the components and the usual methods of expressing concentrations (for example, weight percent, which is one of the overwhelmingly common expressions used, especially in NIR spectroscopy). If a calibration is generated wherein the weight percent is the unit for concentration, then as the amount of analyte departs from the region where the calibration was performed, the weight percent will increasingly deviate from the value predicated on a constant linear relationship between that measure and the volume percent scaled by the algorithm to best match the values given by the analyst in the calibration range. Similarly, we know that samples with a constant volume percentage of analyte may very well exhibit different weight percentages. This behavior is shown in Figures 7a, 8a, and 9a in the September installment of "Chemometrics in Spectroscopy" (24), where weight percent values are plotted against the spectroscopic (CLS) values. Not all of the components exhibit noticeable nonlinearity, but dichloromethane (Figure 8a) and n-heptane (Figure 9a) clearly do. Thus, we can see that those components could not be extrapolated very well. On the other hand, we see in Figures 7b–9b from the same installment, where the volume percent values are plotted against the spectroscopic values, that the data fall on a straight line. There should be little problem in extrapolating a calibration based on one part of the range, to another part of the range for any of those calibrations.
Sources of Overfitting
We have described the mechanism whereby smaller and smaller amounts of nonlinearity in the use of weight percent values for standard calibration are accommodated by the calibration methodology, as more and more PLS or PCR factors are used. This causes the measures of calibration performance to asymptotically approach the noise level of the measurement, but rarely is that point reached before the calibration has included so many factors that the noise is exaggerated, a condition summarized by the term overfitting. The use of volume percent for specifying the analyte concentration should prevent this from happening. In the absence of the nonlinearities engendered by the use of weight percent, calibration performance should improve rapidly as more factors are included in a calibration model and the performance should quickly and sharply reach the ultimate measurement noise level, at a relatively small number of factors, so that overfitting is prevented from occurring.
Need for Dilute Solutions
Historically, spectroscopic analysis has been considered suitable only for dilute solutions. We believe that the reason for this is the same as the reason spectroscopic analysis has been considered nonextrapolatable: When weight percent values, or other concentration measures not conformable to the volume fractions, are used to specify the concentration, then the nonlinearity of the relationship between different units for concentration do not cause noticeable departures between the measurements made against the different types of units, until the range has increased sufficiently for the discrepancy to have an appreciable effect. For sufficiently dilute solutions this discrepancy always remains unimportant.
Instrumental Differences
Over the years, the difficulties encountered have been ascribed to any of several causes: instrument mismatches because of manufacturing tolerances, the lack of a rigorous physical theory to describe the optical behavior of powdered solids, and, of course, the ever-present and infinitely culpable reference laboratory error. Those effects are, of course, real. Nevertheless, attempts to improve NIR performance by addressing those issues never seemed to solve the problem, leaving at least some of us unsatisfied that we were really addressing the correct problems. We know that there was always a nagging feeling in the back of our minds, that some sort of nonlinearity effect would explain the behavior of the NIR calibration process, but neither we nor anybody else could think of a source or a mechanism whereby that sort of nonlinearity would arise in the spectroscopic data. Lo and behold, now that we've finally pinned it down, we see that in fact it does not arise from the spectroscopic data at all, but in the fundamental physical chemistry of the samples. No wonder we couldn't find a cause in the spectroscopic data!
Selections of Wavelengths and Factors
Selection of wavelengths (for MLR), factors (for PLS or PCR), and data transforms have major effects on the ability to perform quantitative calibrations. As we found out, there is no direct effect of these manipulations of the spectra because the key issue is independent of the spectra, and is affected only by the relations between different units used to express the constituent values. Therefore, the manipulations of the spectral data is at best only indirectly related to the calibration performance, mediated through possible differences in the nonlinearities of the spectral data itself.
"Mysteries" in Calibrations
These findings also explain several "mysteries" in spectroscopic calibrations, as well as also make some predictions, and point to some areas where the theory may be extended:
Mysteries in Spectroscopic Calibration
One of the fundamental assumptions in calibration is that we are dealing with linear (mathematical) systems. This assumption is so ingrained that in some incarnations it even forms parts of the name of the algorithms (for example, linear least squares, linear regression, and linear algebra). In high school algebra, we learned to solve systems of linear equations. When we solved systems of linear equations, the solutions were exact, there were no "excess" pieces of the equations left over, and we never had to account for them. We had shown, long, long ago and far, far away (well, ok, maybe not so far as all that) that under suitable circumstances, systems of spectra could also be solved using systems of linear equations (see pages 3–10 in Principles and Practice of Spectroscopic Calibration [25]). Something else we learned in those same high school algebra courses was that you never need more equations than you have variables. In "real-world" spectroscopic analysis, however, we have encountered something not discussed in high school algebra: data has noise (and other types of error). We have learned that to deal with these random effects we need to use many more samples (that is, more equations) than we have variables (wavelengths at which data is measured) and somehow "split the differences" between them. The preferred method of "splitting the differences" is the use of least squares calculations. In various guises, this procedure provides the best estimate available for what would have been attained if there were no noise or error. Ideally, the use of least squares methods would mimic the purely algebraic approach, providing the same spectroscopic model that would be obtained in a noise free situation, and also demonstrate no further benefit from attempting to use more variables (wavelengths or prinicpal component analysis [PCA] factors) than is needed to obtain a "best" fit to the data. We will discuss this point further, below.
Too Many Factors
It is clear to anyone who has performed an NIR calibration that the ideal situation described in the previous section does not exist in the real world. The theory described above would follow standard statistical practice, and error measures would demonstrate improved calibration performance as long as added mathematical variables in fact accommodate the spectral variations caused by real physical (or chemical) variations. As soon as there were enough variables to explain the variations in the data, which would be the number that was equal to the number of actual physical variables, adding further variables would create no further improvement. Reaching this point would be demonstrated not only by a sudden leveling off of the global error measures (standard error of calibration [SEC], standard error of prediction [SEP], and so on), but also by the fact that new variables would have coefficients that were not statistically significant. In practice, we don't observe that. What is more commonly observed is that the global error measures slowly and more or less continually decrease far beyond the number of real physical variables that are expected to exist in the sample set. It's usually not at all clear when to stop adding factors.
Error Measures
Error measures do not go to the noise floor when the number of factors equal the number of variables. This is closely related to the previous "mystery" and indeed is simply a different manifestation of it. Instead of decreasing rapidly while variables representing real physical effects are being added to the calibration model and then suddenly leveling off, there is a slow and more-or-less continual decrease in the error measures, and it sometimes seems that they will never stop decreasing. If correct units for the analyte are used, then the cutoff point for deciding when to stop adding factors should become more clear cut. The number of factors used should also tend to decrease compared to current practice.
Extensions
Here are some areas where theory may be extended:
- The CLS algorithm is the nearest thing we have to a first-principles "absolute" spectroscopic analytical method; therefore results are obtained without the need for conventional calibration. As we have seen from the work we have done, this seems to hold true. As is common, and expected in science, the work should be confirmed and extended by other scientists, using other mixtures. Will this approach become generally useful as an analytical technique? It's hard to say. Numerous practical problems in implementing it would arise and need to be dealt with for each application. Some of these include avoidance of interactions between the components of the mixture, verifying that Beer's law holds for the samples of interest, determining the appropriate measurement units for solids, powders (especially), and other sample types.
- Without the requirement for conventional calibration methods, reference laboratory values are not needed and consequently reference laboratory error is not an issue. However, residual effects of interactions between components, possible nonlinearities in the instrument, the presence of unsuspected mixture components, the inability to obtain spectra of all the mixture components in their pure state, and so on will create measurement errors. These need to be quantitated before the method can be used as a routine measurement tool.
- CLS results are obtained with zero PLS-PCR factors; therefore "overfitting" in the conventional sense is not possible. Underfitting, however, can occur if the analyst is not aware of all the components present in the mixture. Kramer has discussed this situation (26).
Partial Molal Volumes
The fact that the CLS method is absolute has an interesting corollary, one that allows us to predict a new calculation. As long as all the mixture components and their spectra are known, the coefficient computed for each of the mixture components indicates the fraction of the spectral contribution, as compared to the spectrum of the pure component, which is inherently unity. A given volume of each component will contain a specific amount (that is, number of molecules) of the component, and when it is mixed with the other components in the solution it will contribute the corresponding fraction of the volume (and therefore the number of molecules).
Let's consider an example: If water and ethanol formed an ideal solution, and 0.5 mL of water is mixed with 0.5 mL of ethanol, then in the absence of any partial molal volume effects there would be 1 mL of the mixture and a concentration of 0.5 mL of each component per milliliter of solution. This ideal situation is illustrated in Figure 10a.

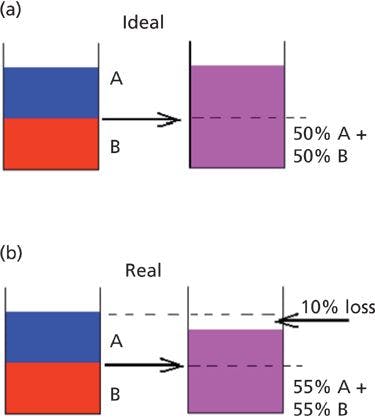
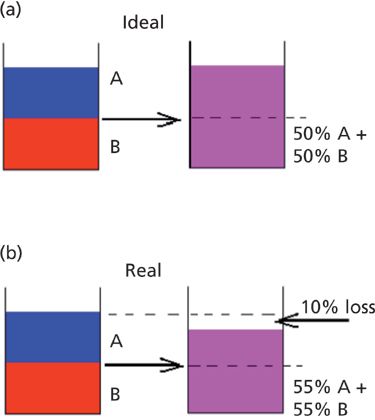
Figure 10: Theoretical effects of partial molal volumes. (a) Hypothetical ideal mixture of water and ethanol, showing no change of volume upon mixing. (b) Exaggerated hypothetical real mixture showing reduction in volume upon mixing.
If, upon mixing, the volume of the solution is less than the sum of the volumes of the components, then that amount of shrinkage, known as the partial molal volume, will pack that same number of molecules into a smaller volume. In reality, of course, water and ethanol are known to exhibit some partial molal effects, on the order of roughly 5% - that is, 0.5 mL of water plus 0.5 mL of alcohol produce only 0.95 mL of solution. This situation is shown, somewhat exaggerated, in Figure 10b.
Consequently, the concentration of each component would be 0.5/0.95 = 0.526 mL/mL of mixture (or 0.55 mL/mL of mixture in the hypothetical exaggerated case shown in Figure 10b). Thus, assuming no change in the spectrum of each component because of the interactions, we would expect the spectrum of each component to have more absorbance than it would have from considerations of only the calculated volume fraction, based purely on the effect of packing more molecules into a given volume. The increased absorbance would translate into correspondingly increased CLS coefficients since they are based on the comparison with the spectrum of the pure components. For the exaggerated hypothetical mixture, we would expect the sum of the coefficients to be 0.55 + 0.55 = 1.0/0.9 = 1.1. How can this be so? Since the volume of the mixture has decreased compared to the pure materials, more molecules are packed into a given volume of the mixture than theory would suggest, giving rise to what would otherwise seem to be an "impossible" concentration. This calculation could thus possibly form the spectroscopic basis for determining partial molal volumes of mixtures.
References
(1) H. Mark and J. Workman, Spectroscopy25(5), 16–21 (2010).
(2) H. Mark and J. Workman, Spectroscopy25(6), 20–25 (2010).
(3) H. Mark and J. Workman, Spectroscopy25(10), 22–31 (2010).
(4) H. Mark and J. Workman, Spectroscopy26(2), 26–33 (2011).
(5) H. Mark and J. Workman, Spectroscopy26(5), 12–22 (2011).
(6) H. Mark and J. Workman, Spectroscopy 26(6), 22–28 (2011).
(7) H. Mark and J. Workman, Spectroscopy26(10), 24–31 (2011).
(8) H. Mark and J. Workman, Spectroscopy27(2), 22–34 (2012).
(9) H. Mark and J. Workman, Spectroscopy27(5), 14–19 (2012).
(10) H. Mark and J. Workman, Spectroscopy27(6), 28–35 (2012).
(11) H. Mark and J. Workman, Spectroscopy27(10), 12–17 (2012).
(12) H. Mark and J. Workman, Spectroscopy 28(2), 24–37 (2013).
(13) H. Mark and J. Workman, Spectroscopy 28(5), 12–21 (2013).
(14) M. Twain, "Quote" in "http://www.quotedb.com/quotes/1097"; (2010)
(15) M. Twain, "Quote" in "http://www.brainyquote.com/quotes/quotes/m/marktwain109624.html" (2010).
(16) W. Rogers, "Quote" in "http://www.brainyquote.com/quotes/quotes/w/willrogers385286.html" (2010).
(17) W. Rogers, "Quote" in "http://www.famousquotesandauthors.com/authors/will_rogers_quotes.html" (2010).
(18) J.D. Ingle and S.R. Crouch, Spectrochemical Analysis (Prentice-Hall; Upper Saddle River, New Jersey, 1988).
(19) J. Workman and H. Mark, Spectroscopy28(6), 28–35 (2013).
(20) J. Workman and H. Mark, Spectroscopy28(10), 24–33 (2013).
(21) J. Workman and H. Mark, Spectroscopy 29(5), 18–27 (2014).
(22) J. Workman and H. Mark, Spectroscopy29(11), 14–21 (2014).
(23) J. Workman and H. Mark, Spectroscopy 22(6), 20–26 (2007).
(24) H. Mark and J. Workman, Spectroscopy 29(9), 26–31 (2014).
(25) H. Mark, Principles and Practice of Spectroscopic Calibration (John Wiley & Sons, New York, 1991).
(26) R. Kramer, Chemometric Techniques for Quantitative Analysis (Marcel Dekker, New York, 1998).
Jerome Workman, Jr. serves on the Editorial Advisory Board of Spectroscopy and is the Executive Vice President of Engineering at Unity Scientific, LLC, in Brookfield, Connecticut. He is also an adjunct professor at U.S. National University in La Jolla, California, and Liberty University in Lynchburg, Virginia. His e-mail address is JWorkman04@gsb.columbia.edu



Jerome Workman, Jr.
Howard Mark serves on the Editorial Advisory Board of Spectroscopy and runs a consulting service, Mark Electronics, in Suffern, New York. He can be reached via e-mail: hlmark@nearinfrared.com



Howard Mark














