What Do You Mean by Good Documentation Practices?
A closer look at the U.S. Pharmacopeial Convention's (USP) draft general chapter <1029> on good documentation practices.



The U.S. Pharmacopeial Convention (USP) has issued a draft general chapter <1029> on good documentation practices for comment. What can we learn from this draft?
In the May 2014 issue of Pharmacopeial Forum, the U.S. Pharmacopeial Convention (USP) issued a draft general chapter on good documentation practices (GDP) (1). Note that this GDP must not be confused with the GDP in the European Union (EU) "good manufacturing practice" (GMP), which stands for good distribution practice in that context. When GDP is used in this column, the "D" will mean documentation.
EU GMP Chapter 4 on Documentation
Before starting to review the United States Pharmacopeia (USP) draft general chapter, let us look at what exists in the regulated world as documentation is the key to compliance with either good laboratory practice (GLP) or GMP regulations. As the section titled "Principle" in EUGMP chapter 4 on documentation states in the first sentence, "Good documentation constitutes an essential part of the quality assurance system and is key to operating in compliance with GMP requirements" (2). This statement can also be applied to GLP or any other quality system such as ISO 17025 or ISO 9001.
Chapter 4 also goes further in describing the types of documents that are possible in a regulated organization:
- Site master file: This describes the GMP activities of a manufacturer. We will not discuss this document in this column.
- Instructions: Includes specifications, protocols, analytical procedures, and standard operating procedures
- Records or reports: Evidence that instructions have been executed
These document types are shown in Figure 1. Of these, the principle (2) describes the two main types of document as follows: "There are two primary types of documentation used to manage and record GMP compliance: instructions (directions, requirements) and records/reports" (2). As shown in Figure 1, the execution of an instruction results in the generation of a record, records, or a report. It is the existence of accurate and reliable records that demonstrate that instructions have been followed and, therefore, compliance with GMP regulations.

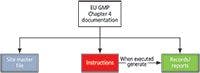
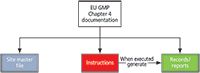
Figure 1: Document types as defined by EU GMP chapter 4. Adapted with permission from reference 2.
Chapter 4 (2) goes into more detail about records and reports:
- Records: Provide evidence of various actions taken to demonstrate compliance with instructions — for example, activities, events, investigations, and, in the case of manufactured batches, a history of each batch of product, including its distribution. Records include the raw data that is used to generate other records. For electronic records, regulated users should define which data are to be used as raw data. All data on which quality decisions are based should at least be defined as raw data.
- Certificates of analysis: Provide a summary of testing results on samples of products or materials (1) together with the evaluation for compliance to a stated specification.
- Reports: Document the conduct of particular exercises, projects, or investigations, together with results, conclusions, and recommendations.
From Chapter 4 (2) we can draw up some basic requirements for documentation for any quality system:
- Say what you do: Have an instruction to document a repetitive task such as an analytical method or a standard operating procedure (SOP)
- Do what you say: Follow the instruction and if there are any deviations from the procedure document them
- Document it: Generate a record to show that the instruction was followed.
This chapter (2) also notes that "The term 'written' means recorded, or documented on media from which data may be rendered in a human readable form." In Chapter 4 speak, a document (instruction or records) does not exist only on paper, it can be any medium as long as it can be converted into a readable format.
Chapter 4 goes further to discuss heterogeneous systems (hybrid systems where there are electronic records with handwritten signatures on paper printouts) and homogeneous systems (electronic signatures and electronic records). The discussion is confused by the mention of raw data in the records section of Chapter 4, shown above. However, the term raw data is not defined in either EU or US GMP as it is a phrase used in GLP (3,4). What does raw data mean in a GMP context — especially as raw data can create other records? The GMP regulations are silent on this subject.
USP Draft <1029> on GDP
Having set the scene, we can now turn to the subject of this column and review the draft USP general chapter on good documentation practices. In overview, there is a preamble outlining the rationale for writing this general chapter and then the chapter itself is divided into the following sections:
- Introduction with scope and purpose
- Principles of good documentation
- Data collection and recording
- Different types of GMP documents
I will discuss each section in the sequence as it appears in the draft general chapter.
Preamble Problems
Our first problem comes in the preamble for the general chapter, which states "This chapter was created to address a need for descriptions of what constitutes good documentation, for example, records of all types that are clear, accurate, and complete." OK, so far so good. However, then comes "These records may include protocols, procedures, reports, and raw data" — now can you see the problem? In our earlier discussion on Chapter 4 documentation (2) and shown in Figure 1, there is a clear demarcation between instructions and records. However, in draft <1029> this distinction is blurred and both instructions and records are intermingled into generic "records." This is wrong and very misleading
Purpose and Scope
There are two paragraphs covering the scope and purpose of the general chapter in the introduction. The purpose statement is relatively straightforward: It aims to describe the underlying principles for documenting GMP activities and to be able to reconstruct what activities were performed, by whom, and when. When we come to the scope of the document we run into another problem. The scope mentions that paper and electronic records can consist of raw data. Ah, the term "raw data" rises from the deep again! Just as helpfully as EU GMP Chapter 4, the draft general chapter fails to define what raw data are. This is really helpful.
Principles of Good Documentation
This section is mainly a bulleted list of requirements for good documentation that need to be interpreted for the media on which the records are recorded. However, when reading the bulleted points it is very clear that the focus of this general chapter and the primary record medium is paper. There is only implicit attention paid to hybrid and electronic systems, but these are the majority of record generating systems in a regulated laboratory. The all-encompassing statement that "Controls should be in place to protect the integrity of the records" is rather inadequate given the industrial-scale falsification and fraud conducted by some companies in the past decade. For more detail on data integrity issues involving chromatography data systems, see my upcoming installment of the "Questions of Quality" column in LCGC Europe (5).
One of the requirements in the draft general chapter is that "short-hand notations are not allowed" — obviously the author of this requirement has not visited a contract laboratory recently. In many contract research organization (CRO) laboratories there is a laminated sheet with change codes posted on the walls throughout the laboratory that detail all the reasons for change with a corresponding number to use when making a correction to a record. The use of these codes is under the control of an SOP. However, in the next section of <1029> is a statement that you can use predefined correction codes, for example "WD" for wrong date. A bad day at the editorial office, perhaps?
Data Collection and Recording
In the section on data collection and recording we reach the nadir of this draft general chapter. The initial section lists four types of documentation, but there is no consideration of hybrid systems; it merely mentions instrument printouts. Ah ha, you may exclaim but it does — there is a mention that states that these four formats ". . . are not limited to." Yes, that is correct. However, as the majority of instruments used in laboratories today are hybrid systems it behooves a body, such as the USP, to include them in such a list. Furthermore, it must state how to control all the records generated by such systems: These records include both the signed paper printouts as well as the underlying electronic records together with the linkage between them. The draft chapter is silent in this respect.
We slide further into the mire as the section then discusses how to record paper records and then describes what you should do for recording and reviewing these paper records. However, the section just adds a bald statement that electronic records have to comply with 21 CFR 11. The focus on paper as the primary record is so last century. Laboratories have moved on from paper. To avoid discussing hybrid and fully electronic systems and the associated records is totally negligent and unprofessional, in my view.
What is required is a discussion about data and the associated metadata for electronic records for both hybrid and electronic systems. What, you may ask, are metadata? The simplest answer is data about data, which is about as useful as a chocolate teapot. A better response is additional data that puts the main data element in context. Let me give you an example:
- You have a spectrum generated by a spectrometer and saved as a file named ABC123. You have a printout of this file and on the printout there is a link to the underlying file. Both the printout of the file and the file itself are your data.
- However these data are useless as there is no context. What is the analysis? What is the spectrum? Who created it? Is there a reference spectrum? Therefore, we need more data to put the spectrum into context and understand why it was generated; this is the role of metadata.
- Additional metadata are required to put the data into context. For example, sample information sufficient to uniquely identity the sample such as study, batch or lot number, or laboratory sample number; instrument identity; method data that defines the instrument parameters used to acquire the data; analysis data for how the file was interpreted or analyzed to obtain the printout; reference spectrum or library reference used in any comparison or calculations; any calculations performed on data derived from the spectrum or spectra; specifications, if applicable, together with the conclusion; audit trail data: who created the file and when this was done plus any further manipulation of the data. In addition, any changes to instrument or analysis settings during the work and the person who made the changes.
Furthermore, there is no mention in the draft <1029> of the criteria for ensuring integrity of records: ALCOA+ criteria defined by regulatory authorities. I discussed these in a "Questions of Quality" column in LCGC Europe on a discussion of fat finger, falsification, and fraud (6).
- Attributable: Who acquired the data or performed an action and when?
- Legible: Can you read the data and any laboratory notebook entries?
- Contemporaneous: Documented at the time of the activity
- Original: Written printout or observation or a certified copy thereof
- Accurate: No errors or editing without documented amendments
- Complete: All data including any repeat or reanalysis performed on the sample
- Consistent: All elements of the analysis such as the sequence of events follow on and dates or time stamped in expected sequence
- Enduring: Not recorded on the back of envelopes, cigarette packets, post-it notes, or the sleeves of a laboratory coat, but in laboratory notebooks or electronically by the electronic lab notebook (ELN) and laboratory information management system (LIMS) used in the laboratory
- Available: Accessible for review and audit or inspection over the lifetime of the record
Different Types of GMP Records
You may recall that a camel is often said to be an animal designed by a committee. So too is the USP documentation camel — the draft <1029> does not distinguish between instructions and records or reports all are listed in no apparent or local order. Perhaps the writers drew out of a hat similar to a lucky dip. The document types are listed below in the order that they are presented in the draft general chapter to which I have added the document type in parentheses:
- Laboratory records (Records)
- Equipment related documentation (Records)
- Investigations and deviations (Records)
- Batch records (Records)
- Certificates of analysis (Reports)
- Standard operating procedures (Instruction)
- Protocols and reports (Instructions and Records)
- Analytical methods (Instructions)
- Training documentation (Records)
- Retention of records (Instruction)
Figure 1 shows an innate logic: When instructions are executed, records are generated. This is not apparent from this <1029> list above. What <1029> should have done is placed the instructions first and the records second, such as shown in Table I.

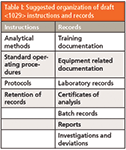
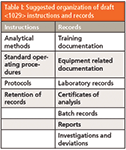
Table I: Suggested organization of draft instructions and records
Nowhere in this section is there mention of the type and nature of the records required to underpin each described type. For example, for laboratory records there is an all-encompassing "measurements," followed by "formulae and calculations," and ending up with "results and conclusions."
What Is Right with Draft <1029>?
It would be unfair of me to just list the negative aspects of the draft USP <1029>, because it provides a good overview of how to record regulated activities on paper. However, it does not mention that pencil and typewriter correction fluid should never be used for documenting GMP activities.
Omissions from Draft <1029>
The omissions are far more serious, in my view:
- Mixing instructions and records is confusing and misleading.
- It fails to define "raw data."
- Its overwhelming focus on paper is wrong and outdated.
- It fails to provide adequate focus on hybrid and electronic systems.
- It fails to provide guidance on the underlying records required to support the record types listed.
Summary
In reviewing the draft USP <1029> general chapter on good documentation practices, this question crossed my mind: Why was this general chapter ever written? What concerns me is that USP is offering training webinars now based on a poorly drafted general chapter. Are pharmaceutical companies and other regulated organizations so incompetent that they cannot provide training in such a basic subject?
References
(1) In-Process Revision <1029> "Good Documentation Guidelines (New)," Pharmacopeial Forum 40(3), May–June (2014), www.usp.org.
(2) European Commission Health and Consumers Directorate-General, EudraLex: The Rules Governing Medicinal Products in the European Union. Volume 4, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Brussels, Belgium, 2011), GMP Chapter 4, Documentation.
(3) Principles of Good Laboratory Practice (Organisation of Economic Co-operation and Development, Paris, 1998).
(4) Good Laboratory Practice for Non-Clinical Studies, 21 CFR 58, (U.S. Government Printing Office, Washington, D.C.,1978).
(5) R.D. McDowall, LCGC Europe, in press (2014).
(6) R.D. McDowall, LCGC Europe 25(4), 194–200 (2012).
R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com



R.D. McDowall














