What Exactly Are Raw Data?
Raw data is a term that is often used in both good manufacturing practice (GMP) and good laboratory practice (GLP) laboratories but can create confusion and misunderstanding. What exactly does it mean and what records are within the scope of the term?
Raw data is a term that is often used in both good manufacturing practice (GMP) and good laboratory practice (GLP) laboratories but can create confusion and misunderstanding. What exactly does it mean and what records are within the scope of the term?
If you want to start a scientific argument with colleagues working in regulated laboratories you can always ask the question, what exactly are raw data? Now it depends which side of the good laboratory practice (GLP) or good manufacturing practice (GMP) fence you happen to be sitting, so we will start with the easy answer first:
- From a GLP perspective the answer will usually be “original observations” given in a firm voice and with plenty of conviction
- In contrast, the GMP answer (usually from a European) will be more tentative, unsure and vague, such as “raw data are used to create other records.” What a succinct answer!
The problem is that if the term raw datais not fully understood it can lead to poor decision making and regulatory noncompliance. This problem is compounded by the failure of some regulatory bodies to define the term or even provide guidance on such an important subject. In this column, we delve into the subject and explore what raw data actually means for a spectroscopic computerized system in today’s regulated GLP and GMP environments.
In the Beginning . . .
Raw data as a regulated term first saw the light of day with the United States Food and Drug Administration’s (FDA’s) Good Laboratory Practice regulations in 1978 (1), where the term is defined in section 21 CFR58.3(k) as follows:
- “Raw data means any laboratory worksheets, records, memoranda, notes, or exact copies thereof, that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study.
- “In the event that exact transcripts of raw data have been prepared (for example, tapes which have been transcribed verbatim, dated, and verified accurate by signature), the exact copy or exact transcript may be substituted for the original source as raw data.
- “Raw data may include photographs, microfilm or microfiche copies, computer printouts, magnetic media, including dictated observations, and recorded data from automated instruments.”
In the regulation there is just a single paragraph, but I have added the bullet points above to aid reading and understanding. You can see how up to date the regulations are when there are references to microfilm and microfiche. However, the first bullet point contains the beginning of the term original observations. Hence, the certainty of any individual who has worked in a GLP environment responds with this term when asked about what raw data are. When framed in the context of spectroscopic computerized system-for example, ultraviolet (UV), near infrared (NIR), Fourier transform infrared (FT-IR), mass spectrometry (MS), and nuclear magnetic resonance (NMR)-the first thought is to focus on the sample file generated from the sample during the course of analysis. These are my raw data. From this misconception, the current problems that we have with data integrity begin.
The issue from many analytical scientists working in GLP regulated laboratories is that they never read the regulations as they pertain to their work. The regulations have been interpreted for them by the great and the good, and handed down like tablets of stone to the laboratory staff to implement and follow. For the raw data debate, the training emphases that original observations must be captured, secured, and protected.
However, this is the time to use one of the corollaries of Murphy’s law, Cahn’s axiom, which states: When all else fails read the manual, standard operating procedure (SOP), or regulation.
Look back to the definition of raw data. Yes, it talks about original observations. But there is more, much more.
“Raw data means . . . records . . . that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study” (1).
Do you see the problems? It is not just original observations but also “activities.” OK, so now we have established that there are more records that could be included. However, the killer line is the one everybody forgets: “necessary for the reconstruction and evaluation of the report for that study.” We will return to this topic when I have finished looking at raw data from a GMP perspective.
Later, Much Later in Europe . . .
Let us move the clock forward from 1978 to 2011 when the European Union (EU) issued an updated version of EU GMP Chapter 4 on documentation (2). Unlike the United States (US) GMP where documentation requirements are spread throughout the regulation like honey on a slice of bread, chapter 4 has the main requirements for documentation in a single location and can be quite explicit in expectations for specifications, instructions, and records. In the Principle of the chapter, under the subject of records we have the following regulatory requirements for records (2):
- “Provide evidence of various actions taken to demonstrate compliance with instructions, [for example,] . . . manufactured batches a history of each batch of product, . . .
- “Records include the raw data which [are] used to generate other records.
- “For electronic records regulated users should define which data are to be used as raw data.
- “At least, all data on which quality decisions are based should be defined as raw data.”
Trivia Quiz Time
- Question: Where is definition of raw data contained in EU GMP? Answer: Nowhere!
- Question: What is the definition of raw data in the US GMP? Answer: There isn’t one!
The problem is when a new term is introduced into a regulated environment there should be a definition of that term so that organizations can interpret and apply it to their processes and systems. However, EU regulators have failed to provide the definition of raw data to enable the industry to begin any interpretation.
Also, you’ll note that the fourth bullet point above contains the ever popular “at least” phrase. What do inspectors interpret this as? This is the minimum, but we would expect more. How does the industry interpret this phrase? This is all we will do. Life is beautiful (at least for consultants).
7th Cavalry to the Rescue?
You will remember at the start of this column that the FDA’s original definition of raw data dates from 1978. Now galloping over the horizon to help the raw data debate is the FDA. I don’t believe I’ve just written that last sentence! Recently, the FDA published their proposals for “Good Laboratory Practice for Nonclinical Studies” (3). This is the first revision of the GLP regulations in nearly 40 years and is based on a comprehensive quality system approach-perhaps the working title of “GLP Quality System” is a bit of a clue.
Rather than turn this column into a review of the proposed regulation, I want to cherry pick one item: the revised definition of raw data. One of the aims of revising the regulation was to address the impact of computerized systems on nonclinical studies. Consequently, the proposal has modified the current definition of raw data in §58.3(k) to address copying requirements, computerized systems, and to include the pathology report. The proposed raw data definition reads as follows (3):
Raw data means all original nonclinical laboratory study records and documentation or exact copies that maintain the original intent and meaning and are made according to the person’s certified copy procedures.
Raw data includes any laboratory worksheets, correspondence, notes, and other documentation (regardless of capture medium) that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study.
Raw data also includes the signed and dated pathology report.
What the FDA has done, by adding other documentation regardless of capture medium and copying, is to eliminate the examples in the original definition we saw earlier. By eliminating the examples, it takes away the media that make the current definition appear so out of date now. The specific inclusion of “the signed and dated pathology report” to what is considered raw data changes the definition of raw data from mere “original observations” to emphasize the whole process from analysis to reporting that is included under the term raw data.
Extracting Principles for GxP Raw Data
What does this mean in practice? How can we interpret raw data for both GLP and GMP laboratories? Let us look at either the proposed or current GLP definition of raw data.
Let’s begin with original observations. How do we make the original observation for a spectroscopic system? We need
- a sampling plan (GMP) or study protocol (GLP) that documents how samples will be taken, stored, and transported,
- a sample with relevant information: identity, study, batch, or lot number, analysis request, and so forth,
- an appropriate and qualified spectrometer,
- an appropriate and validated method including the preparation of the sample for presentation to the instrument,
- reference standards or a library (if we are using the spectrometer for identification), and
- qualified staff to perform the work.
From these prerequisites, the analysis is undertaken and one or more files will be generated and saved by the instrument. These are the first part of raw data. No, not just the data files themselves, but all the other associated contextual metadata that must be linked together to support the generated files containing the identity of the instrument and method used, the analyst performing the work, date and time stamps on the files, audit trail entries, and so forth.
An Interlude for Recap
Returning to the current definition of raw data (in fact, it does not make any difference if we used the proposed definition), it means (3)
any laboratory worksheets, records, memoranda, notes, or exact copies thereof, that are the result of original observations and activities of a nonclinical laboratory study and are necessary for the reconstruction and evaluation of the report of that study
What this means is that the interpretation of the original observation and any calculations or data transformations to obtain the results presented in the report of the work must be considered as raw data. Moreover, any interpretation, calculations, or transformations must be transparent, traceable, and understandable. Does this sound familiar? Does this ring any data integrity bells? Note that the discussion so far has not mentioned data integrity, it comes from the 1978 definition of raw data that is commonly misunderstood. This also includes the army of spreadsheets that many laboratories use for handling and transforming data. Raw data covers the whole analytical process from sample to report.
Continuing the Raw Data Journey
Because we have only acquired the original data files, we now need to interpret the data in accordance with the analytical procedure that we are using-for example,
- comparison with a reference material or standard for confirmation of identity,
- identification using a spectral library and chemometrics,
- identification of an unknown by interpretation of the spectra generated, and
- quantification of the analyte using a calibrated curve.
These steps will generate more records of the work that would include laboratory notebook entries, completed blank or template forms (4), together with further contextual metadata, such as the identity of the library or reference standards, the person who carried out the work, the date and time of work, and audit trail entries.
At the completion of the analytical work, a draft report can be generated for review by a second person reviewer. This step may result in changes required because of, say, typographical errors or the misinterpretation of a spectrum, which will result in more metadata and possibly more files and paper being created. At the end of the process, a final approved report is available. From the new FDA definition (3), this report is explicitly part of the raw data. However, the report is also implicitly part of the raw data under the current GLP definition (1) that nobody bothers to read.
Now we have a better understanding of what constitutes raw data: all files including contextual metadata, records of any sampling and sample preparation, laboratory notebook entries, instrument log book entries for the analysis, spreadsheets, and printouts generated from the sample to the report. Simple!
Visualizing What Raw Data Mean
To see what this means look at Figure 1, which shows what constitutes raw data for a GLP and GMP spectroscopic analysis. Please understand that this chart is a generic representation and aims to present both qualitative and quantitative analysis-to focus one or the other, a little interpretation is required by the reader. This figure is derived from a recent publication by Chris Burgess and myself in my “Questions of Quality” column in LCGC Europe about primary analytical record (5), which is worth reading for additional information on this subject.

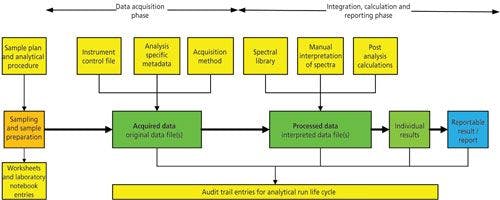
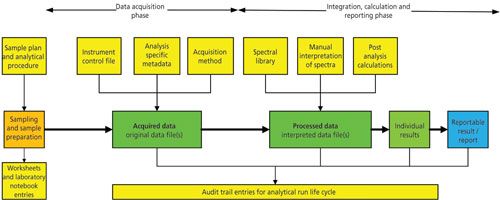
Figure 1: Representative raw data for GLP and GMP spectroscopic analysis.
You’ll see from Figure 1 that the raw data trail starts from the sampling through the sample preparation with work documented in laboratory notebooks, worksheets for which there is accountability (4,6,7) before the sample is presented to the instrument. If we are dealing with identity testing and all that occurs is that the analyst is putting a probe into a drum in a warehouse, then this stage is minimal or omitted altogether. The next stage in the figure is the actual analysis where the right method to control the instrument and acquire the data is used along with sample specific information that is entered to uniquely identify the analysis. Of course, the spectroscopist has logged on and all work including correction of typographical errors is recorded in the audit trail.
Next, the sample spectrum is interpreted by a variety of ways:
- Fit against a composite spectrum in a spectra library for identification (the name of the library and version is part of the metadata supporting the raw data)
- Interpretation of spectra for structure elucidation, here there may be notes in a laboratory notebook associated with the thinking associated with the interpretation that form part of the raw data for the analysis
- Quantification of analytes via calibrated curves or comparison with reference standards depending on the spectroscopic technique being used. Here, we can find that these calculations could be carried out in the instrument data system or a spreadsheet. It is all part of life’s rich raw data tapestry.
Finally, the reportable result will be generated either in the data system or outside it and the report will be written. As noted above in the GLP definitions of raw data (1,3), the report itself is part of the raw data for the work.
Summary: Raw Data Equals Complete Data
In this column, I have discussed that raw data are more than just original observations. The term includes all records created from sampling to reporting and that all stages of the process should be transparent. It also means that an auditor or reviewer can track back from a result in the report to the original observations or forward from the sample to a result in the report.
What should also be apparent to you, as we have done through this discussion, is the similarity between raw data in a GLP context and complete data for GMP as per 21 CFR 211.194(a) (8). In my view, the two terms are equivalent and mean the same thing regardless of the GxP discipline that one is working to. Quod erat demonstrandum?
Acknowledgments
I would like to thank Lorrie Schuessler for the helpful comments made reviewing this article.
References
- Current Good Laboratory Practice for Non-Clinical Laboratory Studies, 21 CFR clause 58 (U.S. Government Printing Office, Washington, DC, 1978).
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 4 Documentation (European Commission, Brussels 2011).
- Current Good Laboratory Practice for Nonclinical Laboratory Studies, 21 CFR Parts 16 and 58, Proposed Rule, Federal Register81(164), 58342–58380 (2016).
- C. Burgess and R.D. McDowall, LCGC Europe29(9), 498–504 (2016).
- C. Burgess and R.D. McDowall, LCGC Europe28(11), 621–626 (2015).
- US Food and Drug Administration, Inspection of Pharmaceutical Quality Control Laboratories (FDA, Rockville, Maryland, 1993).
- US Food and Drug Administration, FDA Draft Gudance for Industry Data Integrity and Compliance with cGMP (FDA, Silver Spring, Maryland, 2016).
- Current Good Manufacturing Practice for Finished Pharmaceutical Products, 21 CFR Part 211 (U.S. Government Printing Office, Washington, DC, 2008).



R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, as well as the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@UBM.com















