Guess Who’s Coming to Inspect R&D?
A new revision to the U.S. Food and Drug Administration (FDA)’s Compliance Program Guide for Pre-Approval Inspections (PAIs) has added a fourth objective: Commitment to Quality in Pharmaceutical Development. PAIs are going to be very interesting now.....
Spectroscopists usually consider chromatography as sample cleanup for their analysis. However, all spectroscopists working in regulated laboratories are living with the fallout of the 2005 fraud perpetrated by the chromatographers at Able Laboratories. The problem was that the Food and Drug Administration (FDA) had carried out seven pre-approval inspections (PAIs) of Able Laboratories, and they had not found any compliance problems until a whistleblower informed them of the compliance issues.
After a two-month inspection by four inspectors, a 483 was issued (1), and the FDA withdrew all product licenses that triggered a recall of over 3100 batches, resulting in Able’s bankruptcy.
Dealing with the Able Labs Fallout
The problem for the FDA was that inspectors focused only on paper printouts—not electronic records during the seven PAIs. Reading the 483, you’ll see how the chromatography data system audit trail aided the inspectors (1). The FDA’s response was to issue a revision of Compliance Program Guide 7346.832 to become effective in 2012 after inspector training. This had three objectives:
- Readiness for Commercial Manufacture
- Conformance with Application
- Data Integrity Audit
Although data integrity runs through all three, it is the third objective where
inspectors should look to see if there was a laboratory data integrity problem.
In 2019, the Compliance Program Guide (CPG) was updated (3), with more emphasis in Objective 3 on laboratory data integrity violations that had been identified since 2010, and this was discussed in a previous “Focus on Quality” column (4). During a PAI, there will be an inspection focus on research and development (R&D) with the documentation and data used to support the product license application inspected under Objectives 2 and 3. Technology transfer to Quality Control is also within scope, as well as any analysis performed for registration purposes.
Guess What? Another CPG Version!
Living in London, we have an expression—”Wait all day for a bus, and three turn up at once.” Here’s the FDA equivalent: in October 2022, the FDA issued another update to CPG 7346.832 (5). Connoisseurs of FDA know that the time between the issue of a draft and final (or more likely another draft) guidance document can be glacial, but three final versions of CPG 7346.832 in 12 years qualifies for the bus comment.
New Objective 4: Quality in Pharmaceutical Development
As mentioned in the abstract, the main change in the latest version is the addition of a fourth objective to CPG 7346.832: Commitment to Quality in Pharmaceutical Development. “Pharm Development” and “Quality” mentioned in the same sentence? Is this a contradiction?
Objective 4 is defined as:
Assess the pharmaceutical development program by evaluating the extent to which it is supported, defined, managed and continually assessed for its effectiveness as well as its use in supporting continual improvement of the Pharmaceutical Quality System (PQS) (5).
What does this mean in practice? The PQS, pharmaceutical development, and continual improvement are mentioned. However, this is within the context of the overall CPG, where one overarching aim is to ensure that data in a regulatory submission is accurate, complete, and consistent, and not been cherry picked. To put this in context, we need to consider the Data Integrity Model (6,7), shown in Figure 1.


Figure 1: A laboratory portion of a data integrity model.
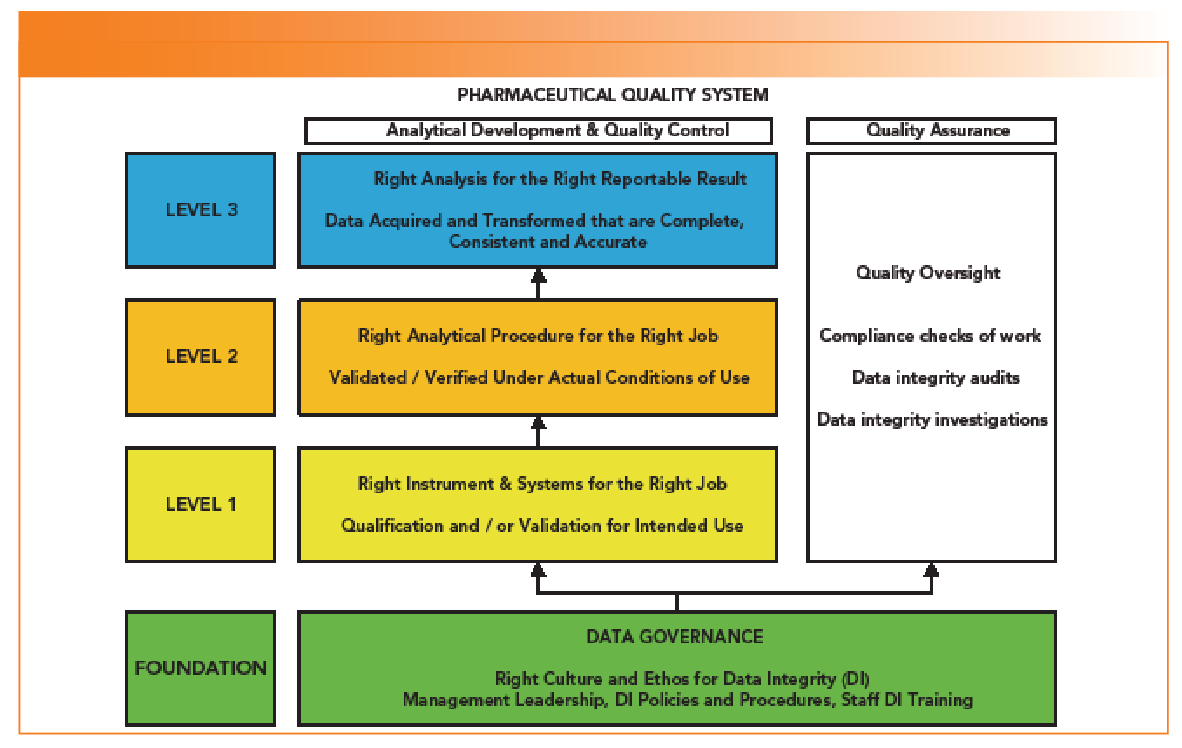
Data Integrity Model and Data Governance
An earlier “Focus on Quality” column, and related publication, discussed a data integrity model within a Pharmaceutical Quality System (PQS) that consisted of a foundation and three levels involving qualification and validation of instruments and systems, validation of methods and application of all to the analysis of samples (6,7). In addition, there must be QA oversight of all work (Figure 1). Objective 4 can be mapped to the foundation level of the model that is essentially data governance:
- Management leadership
- Open culture and ethics
- Good documentation practice
- Data integrity procedures with effective training
The goal is to find the right people and approach to do the job. If an organization can’t get these fundamentals right, we end up with another Able Laboratories.
What Will the FDA Be Inspecting?
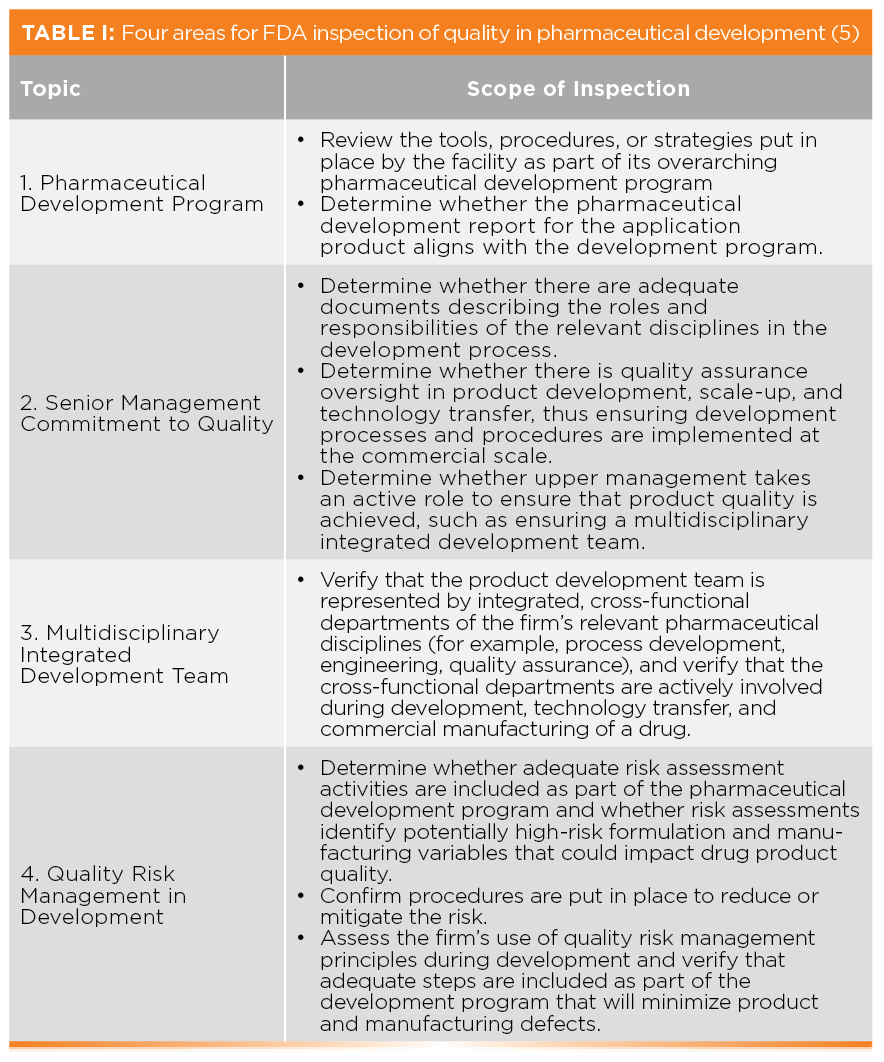
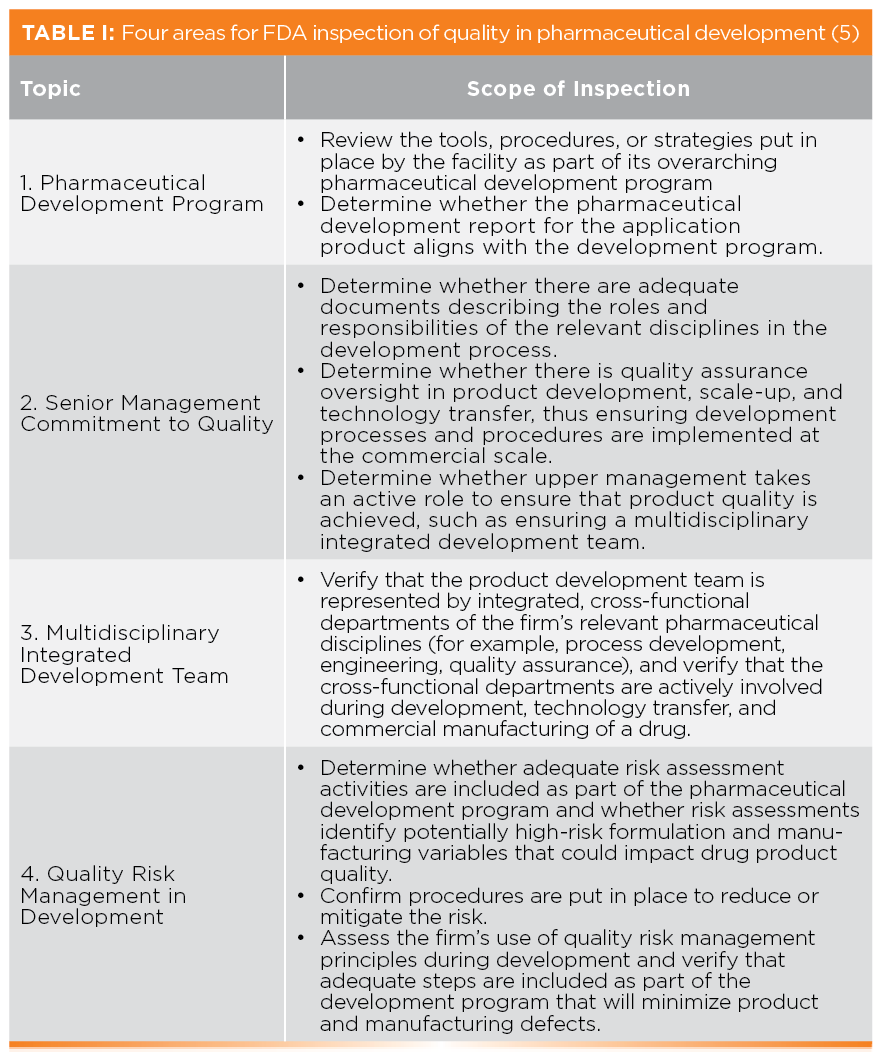
The CPG breaks down Objective 4 down into four areas that are indicative of an organization’s commitment to quality in pharmaceutical development and shown in Table I. The four areas are:
- Pharmaceutical Development Program
- Senior Management Commitment to Quality
- Multidisciplinary Integrated Development Team
- Quality Risk Management in Development
Objective 4 reflects the integration of the principles of ICH Q8(R2) on Pharmaceutical Development (8) and the recently updated ICH Q9(R1) for Quality Risk Management (9) into FDA PAIs. This is a good example why any regulated organization must keep current.

ICH Q8(R2) states:
The aim of pharmaceutical development is to design a quality product and its manufacturing process to consistently deliver the intended performance of the product [8].
This means development of a manufacturing process, the product formulation, and all associated analytical procedures covering raw materials, in-process samples, and finished product analysis.
We’ll discuss two aspects of Objective 4 here:
- Role of Senior Management in Quality
- Technology transfer of analytical procedure from Analytical Development to Quality Control.
Senior Management Commitment to Quality
Management leadership in the foundation layer of the Data integrity Model in Figure 1 is the key to data integrity and quality success. What is expected of senior management? From a data integrity perspective, the 2018 FDA guidance is very explicit:
Meaningful and effective strategies should consider the design, operation, and monitoring of systems and controls based on risk to patient, process, and product. Management’s involvement in and influence on these strategies is essential in preventing and correcting conditions that can lead to data integrity problems.
It is the role of management with executive responsibility to create a quality culture where employees understand that data integrity is an organizational core value and employees are encouraged to identify and promptly report data integrity issues. In the absence of management support of a quality culture, quality systems can break down and lead to CGMP non-compliance (10).
This data integrity expectation of FDA is then rolled into the scope and intent of Objective 4 in the CPG, which is then expanded:
Assess the establishment’s ability to develop and manufacture drugs of consistent quality. This includes determining whether an establishment has implemented and follows a development program that applies sound science and principles of material science, engineering, knowledge management, and quality risk management in a holistic manner (5).
Senior management does not escape the responsibility for the PQS under EU GMP Chapter 1.5:
Senior management has the ultimate responsibility to ensure an effective Pharmaceutical Quality System is in place, adequately resourced, and that roles, responsibilities, and authorities are defined, communicated and implemented throughout the organization. Senior management’s leadership and active participation in the Pharmaceutical Quality System is essential. This leadership should ensure the support and commitment of staff at all levels and sites within the organization to the Pharmaceutical Quality System (11).
Thus, senior management can make or break integrity and quality.
At Able Laboratories (1), senior management had an expectation that all product batches would meet specifications—or you’re fired. How’s that for an incentive to bend the rules? Recently, intense management and investor pressure on research staff at vaccine startup Laronde led to data falsification and the inability to replicate experiments, resulting in cancellation of two late-stage research projects (12). An R&D version of Able Laboratories
without the heavy plod of FDA footsteps!
Technology Transfer of Analytical Procedures
For the second area of Objective 4, we need to look at technology transfer. Stage left we have Analytical Development, who are responsible for developing and validating analytical procedures. Stage right we have Quality Control (QC), who have the dubious task of establishing a validated procedure and using it for routine analysis of production batches. Center stage, there is space to build a large wall between the two departments. The building of the wall is dependent on how the two departments communicate for technology transfer. No wall equals much communication; high wall…...
Consider the following:
- Is QC consulted on the development of the procedure?
- How is the analytical procedure communicated? Throw the report over the wall and let QC get on with it? Are there discussions with QC and their involvement in the development and validation?
- Are equivalent instruments available in both departments to ease the transfer?
- Are Analytical Development’s spectrometers qualified to the same parameters used in QC? Indeed, are they qualified at all?
- Are the same data systems used in both Departments?
Why are these questions important? Let us return to the trigger for this column, CPG 7346.832, under Objective 4, states:
…. comprehensive process and product understanding to the extent possible (5).
If there is no collaboration between the two departments, how can this information and knowledge be transferred to QC? Where is the commitment to quality required by Objective 4?
Table II compares the functions of analytical development and quality Control; the key to our discussion is the bottom row with the relationships for transfer and establishment of an analytical procedure in QC.



Technology Transfer and Data Sharing
In an ideal world, technology transfer would be facilitated by using the same spectrometers and software so that the instrument methods can be transferred electronically with spectra. This would give the receiving laboratory more detail than is generally available in a written document. Rarely is it an ideal world, as R&D and Production have different budgets. There are two options for optimal transfer of analytical procedures:
- Share the overall validation between the two departments. The main work is conducted by Analytical Development, but intermediate precision involves QC staff working with their instruments in their laboratories. Both departments are co-signatories of the final validation report.
- A member of QC could work in the originating laboratory to learn and understand the procedure and speed establishment in their own laboratory, simplifying the transfer protocol.
Regardless of the transfer mechanism, how long should Analytical Development support QC after the procedure has been transferred? QC analysts may need access to development records, in case of situations where a procedure may need to be modified in the future. Ongoing data sharing and collaboration is critical for success.
Frequency of Objective 4 Inspection
Helpfully, on page 14 of the CPG, the frequency of inspecting Objective 4 is noted (5):
- On the initial PAI
- Periodically in subsequent PAIs, with the frequency based on risk
- In addition, when there have been major changes to the quality system, management team or corporate structure (at the rate of change and merger in some organizations this can mean every PAI).
The depth of coverage during an inspection will vary based on the risk and application specific issues identified. To summarize, if you have been good boys and girls in a stable environment, then you’ll see less of an inspector for this objective. Otherwise, you could be on first name terms….
Summary
The publication of a new FDA compliance policy for PAIs brings a new area for inspection: quality in pharmaceutical development. Senior management are responsible for ensuring the culture and ethos in R&D, and should not apply pressure on staff or data integrity suffers. Communication and involvement in analytical procedure validation and transfer between Analytical Development and QC is critical for success.
References
(1) Able Laboratories Form 483 Observations. 2005; Available from: https://www.fda.gov/media/70711/download
(2) Compliance Program Guide CPG 7346.832 Pre-Approval Inspections. 2010, Food and Drug Adminsitration: Silver Spring, Maryland.
(3) Compliance Program Guide CPG 7346.832 Pre-Approval Inspections. 2019, Food and Drug Administration: Silver Spring, Maryland.
(4) McDowall, R. D. Do You Understand the FDA’s Updated Approach to Pre-Approval Inspections? Spectroscopy 2019. 34 (12), 14–19.
(5) Compliance Program Guide CPG 7346.832 Pre-Approval Inspections. 2022, Food and Drug Administration: Silver Spring. Maryland.
(6) McDowall, R. D. Understanding the Layers of a Laboratory Data Integrity Model. Spectroscopy 2016, 31 (4), 15–25.
(7) McDowall, R. D. Data Integrity and DataGovernance: Practical Implementation in Regulated Laboratories; Royal Society of Chemistry, 2018.
(8) ICH Q8(R2) Pharmaceutical Development. 2009, International Council on Harmonisation: Geneva, Switzerland.
(9) ICH Q9(R1) Quality Risk Management. 2023, International Council for Harmonisation: Geneva, Switzerland.
(10) FDA Guidance for Industry Data Integrity and Compliance With Drug CGMP Questions and Answers 2018, Food and Drug Administration: Silver Spring, Maryland.
(11) EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 1 Pharmaceutical Quality System. 2013, European Commission: Brussels, Belgium.
(12) DeAngelis, A.; Cross, R. The Inside Story of How Data Integrity Issues Roiled a Biotech Seen as “Moderna 2.0.” 2023. Available from: https://www.statnews.com/2023/06/12/laronde-data-integrity-issues-biotech/ (accessed 2023-08-01)


R.D. McDowall is the director of R.D. McDowall Limited and the editor of the “Questions of Quality” column for LCGC Europe, Spectroscopy’s sister magazine. Direct correspondence to: SpectroscopyEdit@MMHGroup.com ●








Synthesizing Synthetic Oligonucleotides: An Interview with the CEO of Oligo Factory
February 6th 2024LCGC and Spectroscopy Editor Patrick Lavery spoke with Oligo Factory CEO Chris Boggess about the company’s recently attained compliance with Good Manufacturing Practice (GMP) International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Expert Working Group (Q7) guidance and its distinction from Research Use Only (RUO) and International Organization for Standardization (ISO) 13485 designations.






