Validation of Spectrometry Software: Understanding the Role and Content of a Validation Master Plan
Columnist Bob McDowall discusses the role of a validation master plan (VMP) for summarizing a laboratory's approach to computer validation.



The topic of this column is the role and content of a validation master plan (VMP). I want to discuss where it can be used within the analytical laboratory to summarize your approach for validation of computerized systems and qualification of analytical instruments, as well as to provide a list of systems and their current status. We will look at the definition of a VMP, what the regulations say, how to interpret them for a laboratory environment, and finally, look at how we can manage our spectrometry systems and their validation status effectively within this document. Although I will not be discussing this in great detail, the validation master plan must consider instrument qualification as well as the computerized system validation aspects of laboratory equipment as part of an integrated approach to automated instrument qualification and computerized system validation.



R.D. McDowall
Definitions
Before I dive in and start discussing a VMP in depth, I'd like to define the terminology used in this column to ensure that we all understand what I am talking about (I did try to avoid using the phrase on the same page but the temptation was too great so it is sneaked in). Plans are very important in computer validation, as they define what you are going to do and what you will have as evidence of actions. However, there is a terminology issue, as we can have a number of plans:
- Validation plan
- Master validation plan
- Validation master plan
What do these plans really do? The term master validation plan is, in my view, another name for a validation plan and will not be used any further in the column. To use it further only complicates the issue when we should look for simplification. Therefore, for the purposes of this discussion I will focus only on two: validation plan and validation master plan.
Validation plan: This is the controlling document for the validation of a single computerized system. It defines who is involved and what they do (roles and responsibilities) as well as the life cycle phases to be followed and the anticipated documented evidence to be written to support the validation. It is a high-level document that does not detail what testing is to be undertaken, as that is the role of a test plan.
The FDA in Section 4.5 of the General Principles of Software Validation notes, "The software validation process is defined and controlled through the use of a plan. The software validation plan defines "what" is to be accomplished through the software validation effort. Software validation plans are a significant quality system tool. Software validation plans specify areas such as scope, approach, resources, schedules, and the types and extent of activities, tasks, and work items" (1).
VMP: Rather than present my own views, I leave it to the regulators: A VMP is a document that summarizes the firm's overall philosophy, intentions, and approach to be used for establishing performance adequacy (2). In essence, the document can be very wide ranging and summarizes the approach to validation for a site, division, department, and computerized and laboratory systems. As such, the document must have a scope statement to set the boundaries and define what is in scope and what is outside of it. During this column, I'll focus on the analytical-spectroscopy laboratory.
Regulatory Background to the VMP
In the United States, there is no specific requirement for a VMP; however, many inspectors expect an inventory of computerized systems. In contrast, under European Union (EU) GMP Annex 15, there is a regulatory requirement and a regulatory expectation for a VMP (3). The main requirements for computerized systems are defined in EU GMP Annex 11 (4), but because the regulations were written in 1992, there is currently no linkage with Annex 15. However, as Annex 11 is currently under revision, I expect to see the updated Annex 11 include the requirement for a VMP for computerized systems.
EU Annex 15 covers qualification and validation but was derived from an earlier Pharmaceutical Inspection Convention/Pharmaceutical Inspection Cooperation Scheme (PIC/S) document, as we shall see. PIC/S is an organization for the major regulatory agencies in Europe as well as Japan, Canada, and Australia. Why this organization is important in the U.S. is that the FDA is now applying for membership in the organization, whereas previously it had observer status. There are a number of publications produced by PIC/S that are available for download from their web site at www.picscheme.org. One of these has the snappy title of "Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation and Cleaning Validation" (2). This document was issued originally in 1996 but was updated in 2001. It is this document that was used as the basis for Annex 15.
However, in writing Annex 15, the authors butchered the content of the VMP section of the PIC/S document down to a series of bullet points covering half a page rather than the four pages in the source document — but the heart of the document was lost in translation (from English to English?). Therefore, to understand fully the role and content of what a VMP really is and the thought behind its content you should read the original PIC/S document (2).
Why Do You Need a VMP?
The biggest problem when joining a new company or auditing or inspecting one is finding how things get done and what's going on. Computerized system validation and instrument qualification are both multidisciplinary jobs involving various professions and skill sets — a VMP is a way to organize and manage the multiple jobs over several laboratories or departments, or within a facility. Think of the poor soul in QA wondering what the status is of various validation projects going on in the laboratory — here's the solution! The VMP can present the approach, the systems, their current validation status, and the timescales for any planned or ongoing work in a nutshell.
To quote the PIC/S guide (2): The VMP should present an overview of the entire validation operation, its organizational structure, its content and planning. The core of the VMP being the list-inventory of the items to be validated and the planning schedule. In writing a VMP you will help management, quality assurance, laboratory staff, any validation personnel, and inspectors. What you need to do is define the scope of the document: What is in and what is out?
Defining the VMP Scope
This is where you need to be smart but also careful when interpreting the PIC/S document for computerized system validation. The guidance mentions manufacturing and process equipment, so you'll need to define the scope with the slant to the laboratory. Although with such a flexible document, there is nothing to stop you having a laboratory section within it or indeed a VMP for the laboratory.
So the scope of the document will be defined by a number of factors:
- Physical boundaries: site, division, building, department, or laboratories. Indeed, a VMP for some activities such as Part 11 can go across sites.
- Driver for the plan: This can be a manufacturing process, site computerized systems, or laboratory systems (not forgetting your army of spreadsheets!).
- IT infrastructure: This can be included or excluded depending upon the size of the operation. Typically, this is excluded in larger organizations, as it is the responsibility of corporate IT; however, in smaller organizations it can be included within the scope of a VMP. My inclination is to exclude this and have IT take care of the work; however, this must be documented in the exclusions section of the document.
- Defining the qualification, validation, and calibration activities that will contribute to the overall control over these systems.
- All activities covering prospective and retrospective validation and qualification of systems within scope as well as any revalidation and requalification tasks.
Just as important as defining what is under the scope of the VMP is stating what is excluded from the scope of the document. Therefore, there must be a section stating what is excluded from the plan together with a rationale: Systems with no regulatory impact (excluded from validation but listed in the appropriate section of the inventory) and IT infrastructure (if under separate responsibility of the IT group).
A VMP is also a formal document that should be reviewed and released by management and QA. Who signs the document depends upon the scope of the VMP; it could be the site head or a department head. What should be clear is the role of each signatory to the document: Technical author, technical review, compliance review, and approval followed by the document release.
Size of a VMP
Inspectors and auditors are not impressed by a document's size — or thud factor when it hits the desk. Being a suspicious bunch, we like to think there is something hidden, and that motivates a desire to find out what it is. Alternatively, if you have a canny inspector, they'll ask the questions and you'll do the hunting for them! Therefore, what you want is a summary document that should therefore be brief, concise, and clear. It should not repeat information documented elsewhere, but refer to existing documents such as policy documents, standard operating procedures, and validation protocols and reports (2). So you need a section on referenced documents and just a summary of your laboratory's approach to validation and qualification. Got the message?
Sections of a VMP
Let's move from theory to practical: What should a VMP contain? Table I shows the main sections of a VMP that has been adapted from the PIC/S guidance (2) and modified in light of my experience in writing these documents for laboratories and organizations. Where possible, keep the writing short and cross-reference your policies, guidelines, and procedures. Remember the PIC/S guidance: The document should be concise.



Table I: Sections and subheadings of a laboratory validation master plan
Are You in Control?
If one word is needed to summarize what we are trying to achieve through a number of activities such as validation and qualification, it is "control." The question that any inspector would ask is: "Are you in control?" Control, in this context, is a concept that covers your processes, instrumentation, and analyses. The main way to achieve control is through one or more of the following:
- Computer validation
- Instrument qualification
- Calibration of instruments against standards (metrology based)
- Calibration of instrument against the method (system suitability test or equivalent)
- Preventative maintenance
- Change control
- Breakdown maintenance
Of these, the last is only a response to a problem — it's broken, so fix it. The remainder are either planned activities or in-analysis mechanisms to demonstrate that the spectrometer system is under control. Therefore, VMP must reflect what the laboratory will do to achieve this; not in detail, but cross-referencing your key procedures covering all of these areas. We must get a balance between computer validation and the other control mechanisms available to us in the laboratory. Why produce piles of paper if you run a calibration to show the instrument and software are working over the operational range required?
The Inventory: Setup and Maintenance
As noted earlier, the heart of the VMP is the systems inventory (2). Here we'll look at how it must be set up and maintained. From Table I, there should be a listing of high-and low-risk systems and those not requiring validation. OK, that's easy. However, we also need a little more information appended to each system:
Who is responsible for the system (system owner)?
Where is it located (department)?
What does it do (this ideally is a short description of the function of the system)?
What is the validation status (being implemented, validated, upgrading, and so forth)?
More information can be added, depending upon the organization but these are the core requirements for each system.
The Inventory: Updating Problems
The underlying issue with the inventory is that it is dynamic: systems, instruments, and spreadsheets are being added to the inventory or retired and removed from it. Sometimes systems will be upgraded to utilize new functions in the software. The bottom line is that the inventory will change over time and will need to be updated to reflect the current situation. However, in a paper world, this could mean reissuing a VMP every time the inventory needs updating, which is not very practical.
It is better to separate the main sections of the VMP from the inventory so that both are controlled documents but the inventory can be updated separately. This minimizes the paper volume but does not get the laboratory away from updating the document on a regular basis. So, typically, the inventory is updated every 3 or 6 months (or when an inspection is due), which is not ideal because there will always be a disconnect from the inventory and reality, so the pending entrants to the inventory should be kept with the person responsible for maintaining it.
Automating System Risk Assessment with Inventory Update
Wouldn't it be nice if, in an ideal world, we could automate the whole process so that when a system underwent a risk assessment, the VMP inventory could be updated automatically with the system allocated to a high-or low-risk category? Then, when we completed a validation and approved the validation summary report, the inventory would be updated automatically? This is shown diagrammatically in Figure 1; each stage of the risk assessment and validation results in an automatic upgrade of the VMP inventory to save time.

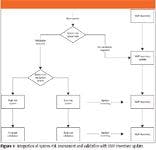

Figure 1
Welcome to the world of computerized systems validation! This is where we have moved from the paper-driven process that characterizes our current approach to computer validation to an electronic environment to speed up the validation of our spectrometer systems. This is the future of computer validation and, of course, will be reflected in the VMP and its inventory.
Summary
I have looked at the role of the validation master plan and the associated inventory as a summary of a laboratory's overall instrumentation qualification and computer validation efforts.
R.D. McDowall is principal of McDowall Consulting and director of R.D. McDowall Limited, and "Questions of Quality" column editor for LCGC Europe, Spectroscopy's sister magazine. Address correspondence to him at 73 Murray Avenue, Bromley, Kent, BR1 3DJ, UK.
References
(1) FDA Guidance for Industry, General Principles of Software Validation, 2002.
(2) PIC/S Guidance, Recommendations on Validation Master Plan, Installation and Operational Qualification, Non-Sterile Process Validation and Cleaning Validation, 2001.
(3) European Union Good Manufacturing Practice, Annex 15.
(4) European Union Good Manufacturing Practice, Annex 11.















