|Articles|July 1, 2018
- Spectroscopy-07-01-2018
- Volume 33
- Issue 7
Enhanced Protein Structural Characterization Using Microfluidic Modulation Spectroscopy
This article introduces a new IR technique, microfluidic modulation spectroscopy (MMS), that is designed to address the needs in biotherapeutics, and presents data from measurements of commercially available proteins.
Advertisement
Measurement and characterization of the secondary structure of proteins are critical in many research applications, especially the formulation and development of biotherapeutics. Traditional analytical tools are not optimized for the demanding requirements of these applications, which include high sensitivity, wide dynamic range, simplified workflow, and high repeatability. This article introduces a new infrared (IR) technique, microfluidic modulation spectroscopy (MMS), that is designed to address these needs, and presents data from measurements of commercially available proteins. The data demonstrate significant increases in sensitivity, dynamic range, and utility for the determination of protein similarity (fingerprinting), quantitation, protein secondary structure, and protein stability and aggregation through thermal and chemical denaturation methods.
Proteins are large molecules with complex three-dimensional (3D) structures that directly influence their efficacy and safety as biotherapeutics. The primary structure of proteins is the sequence of amino acids in the peptide backbone with secondary structures arising from interactions between hydrogen bond donor and acceptor residues associated with repeating peptide units. These interactions promote stabilized folding of the peptide chains principally to form α-helix or β-sheet–strand substructures, the geometry of which is defined by the intermolecular forces, bond angles, and planar configurations involved. However, the secondary and associated higher order structure of proteins is not fixed, but rather is dependent on the localized environment, changing in response to, for example, thermal or chemical stresses. This propensity for instability or conformational change that is often accompanied by aggregation, the process by which proteins start to bind together under different conditions and formulations, is a defining issue in the discovery, formulation, and manufacture of biotherapeutics and biosimilars.
Measuring Protein Structures in Drug Development, Formulation, and Manufacture
Promising biological drug candidates, those that exhibit therapeutic activity and inherent stability, as assessed via simple screening techniques, become subject to increasing levels of structural elucidation as they progress through the pharmaceutical pipeline. Detailed definition of the structure of a drug molecule provides a basis for the identification of structure–function relationships, for understanding the mechanisms by which the drug is efficacious, but beyond this, structural characterization plays a critical role throughout the drug development life cycle. In particular, investigating structural changes is the key to understanding and controlling the factors and mechanisms associated with stability and aggregation.
Defining an optimal formulation and manufacturing route relies on assessing the impact of variables such as pH, excipient choice, processing conditions including temperature and applied shear stress, and storage. Stress-induced structural changes may have significant consequences including a loss of efficacy, and in the worst case present a safety risk, so demonstrating comparability (that successive stages of formulation, manufacture, and storage do not materially impact the structure of the drug) up to the point of administration is essential.
The demonstration of comparability relies on showing that the quality attributes of the drug remain "highly similar and that the existing knowledge is sufficiently predictive to ensure that any differences in quality attributes have no adverse impact upon safety or efficacy of the drug product" (1). The accurate measurement of secondary structure and changes in secondary structure underpin these efforts and are critical activities across the drug life cycle, including in the demonstration of biosimilarity. The use of biosimilars follow-on biotherapeutics with such closely comparable performance to an original drug that they can be used interchangeably, has the potential to drive down health care costs but is highly dependent on the rigorous demonstration of similarity in a wide range of attributes including protein content, activity, and stability. The demonstration of a comparable secondary structure via robust analysis is an efficient way to directly support claims of biosimilarity.
Using Vibrational Spectroscopy for Protein Characterization
Vibrational spectroscopy is an established tool for protein characterization (2), with Fourier transform infrared (FT-IR) wavenumbers in the range 1700–1500 cm-1 routinely used to probe the amide I (1700–1600 cm-1) and amide II (1600–1500 cm-1) bands respectively. The amide I band is associated with the C=O stretch vibration of peptide linkages and quantifies the strength of the carbonyl bond along the protein backbone, which is highly sensitive to changes in secondary structure (3). Indeed, absorption features of the amide I band are well correlated with shifts in hydrogen bonding, dipole-dipole interactions, and geometric orientations in the α-helices, β-sheets, and turns, and with other less prevalent motifs of secondary structure such as random coils (4). The relative amounts of different elements of substructure can therefore be precisely determined by comparing measured amide I spectra with those gathered for proteins with well-characterized secondary structures (5) to provide valuable information for the assessment of, for example, chemical and thermal stability (6), aggregation (7), and biosimilarity (8,9). Notably, IR is particularly sensitive to the β-sheet structures that are prevalent in protein antibody drugs and is one of the very few analytical techniques that can be used to directly monitor aggregation processes because of its ability to measure the intermolecular β-sheet structures associated with aggregate formation (10).
Though the potential of vibrational spectroscopy in protein analysis is widely recognized, current commercial presentations of the technology are not optimally matched with requirements for secondary structure measurement within biotherapeutic development, formulation, and manufacture. As a result, researchers routinely deploy a suite of diverse analytical techniques to study the outlined issues associated with protein structure, with each technique somewhat aligned with the constraints and requirements of different stages of the development cycle. Differential scanning calorimetry (DSC) is routinely used as an early screen for thermal stability, for example, while size-exclusion high performance liquid chromatography (HPLC) can be used to detect aggregation because of its capabilities for molecular size measurement. Neither of these techniques provides detailed structural characterization of the protein. FT-IR and Raman are both used for structural analysis but require sample concentrations of at least 10 mg/mL and 30 mg/mL, respectively (10), with FT-IR working optimally in the 10–150 mg/mL range. This concentration requirement can be a limitation for application in earlier stages of development when sample availability can be an issue. FT-IR also has other well-documented issues including a tendency to exhibit background drift, low sensitivity, and a requirement for close temperature control.
Ultraviolet–circular dichroism (UV-CD), in contrast, is an established tool for structural analysis at more dilute concentrations, typically operating in the range 0.2–2.0 mg/mL (11), but it is unsuitable for direct measurement at high-formulation concentrations. Biotherapeutics are typically delivered intravenously or subcutaneously in liquid formulations, often at relatively high concentrations to minimize administration time within the limitation of having a stable formulation of practical viscosity. Sample dilution complicates measurement and introduces an additional source of analytical variability, and the extrapolation of measured results to higher concentrations is widely recognized as a potential source of error in biopharmaceutical characterization because of the sensitivity of protein structure to its surrounding environment. Furthermore, UV-CD is relatively insensitive to changes in β-sheet structure, especially intermolecular β-sheet structures, compromising its application in the study of aggregation mechanisms.
Introducing Microfluidic Modulation Spectroscopy: High Sensitivity IR Across a Wide Dynamic Range
A new technique called microfluidic modulation spectroscopy (MMS) directly addresses the limitations of current technologies and provides an efficient tool for direct, label-free protein analysis. It uses a tunable mid-infrared quantum cascade laser to generate an optical beam that is 1000 times brighter than those used in conventional FT-IR, enabling the measurement of samples that are substantially more concentrated and the use of simpler detectors with no requirement for nitrogen cooling. The laser is run in continuous wave mode to generate a very high resolution (< 0.001 cm-1 linewidth), low noise beam with minimal stray light that is focused through a microfluidic transmission cell with a relatively long (25 µm) optical pathlength onto a thermoelectrically cooled mercury cadmium tellurium (MCT) detector. This optical configuration delivers high sensitivity measurement over a concentration range of 0.1–200 mg/mL for structural characterization and down to 0.01 mg/mL for protein quantitation, giving MMS a far wider dynamic range than alternative protein characterization techniques.
In an MMS instrument (see Figure 1), the sample (protein-in-buffer) solution and a matching buffer reference stream are introduced into the transmission cell under continuous flow and then rapidly modulated (1–10 Hz) across the laser beam path to produce nearly drift-free, background compensated, differential scans of the amide I band. The complete optical system is sealed and purged with dry air to minimize any interference from atmospheric water vapor, which absorbs across the 2000–1300 cm-1 wavenumber range and can therefore compromise the use of IR spectroscopy for protein characterization. Advanced signal processing technology is the third key element of the instrument and converts the raw spectra into fractional contribution data for specific motifs of secondary structure, providing a structural fingerprint of the protein.
Figure 1: In an MMS instrument the protein sample is rapidly modulated across the laser with a matching water-buffer stream to produce highly sensitive, differential IR scans of the amide I band that provide detailed structural information.
MMS is continuously autoreferencing, eliminating the problems associated with background drift in FT-IR measurements through the use of rapid modulation and the associated generation of a differential measurement. It streamlines the utility of vibrational spectroscopy for biopharmaceutical analysis by combining high repeatability and sensitivity with the wide dynamic range needed to address measurement requirements from development through to quality control, and comparability assessment. The following examples show its application to routinely encountered proteins, demonstrate how performance compares with that of other techniques, and illustrate the value of MMS for protein quantification, structural conformation, the investigation of stability and aggregation, and similarity assessment.
Experimental
A series of experiments was carried out to demonstrate the dynamic range, repeatability, and sensitivity of MMS using a range of commercially available proteins. The specific details relating to the generation of each dataset are given below. All measurements were carried out using a prototype MMS platform (RedShift BioAnalytics Inc.) at a temperature of 21 °C with no cooling of the fluids or measurement cell.
Experiment I: Demonstrating Dynamic Range Through the Measurement of Bovine Serum Albumin Across a Range of Concentrations
Bovine serum albumin (BSA) was obtained from Sigma-Aldrich and prepared in 0.016 M PBS (phosphate buffered saline, pH 7) buffer to generate a concentration series from 0.1 to 200 mg/mL. Measurements were performed under otherwise identical conditions using MMS. A modulation frequency of 1 Hz was applied during measurement and spectra were gathered from 1584 cm-1 to 1714 cm-1 to cover the amide I band.
Experiment II: Assessing Repeatability via Duplicate Measurements of Hen Egg White Lysozyme
Multiple measurements of replicate hen egg white lysozyme (HEWL) samples (Sigma-Aldrich) were performed to assess the repeatability of secondary structure results. The samples were made up to a concentration of 10 mg/mL in water and measured at room temperature. The reference stream used during measurement was water and a modulation frequency of 5 Hz was applied. Again, spectra covered the amide I band.
Experiment III: Comparing the Sensitivity of MMS and FT-IR for the Detection of Structural Impurities by Spiking Immunoglobulin (IgG1) with BSA
To investigate the ability of MMS to detect structural impurities, BSA (a predominantly α-helical protein) was added to a solution of IgG1 (a predominantly β-sheet protein) in known concentrations and the resulting mixtures were then analyzed using the techniques of FT-IR and MMS. Data were gathered for a 100% IgG1 solution and for solutions of 2%, 4%, 6%, 8%, and 10% w/w BSA in IgG1 solutions. The buffer used for all solutions was 10 mM potassium phosphate, pH 7.0, and measurements were carried out at an overall concentration of 20 mg/mL. FT-IR measurements were made using an MCT detector and an attenuated total reflectance (ATR) cell (Bruker). MMS measurements were made using blank buffer as a reference fluid and a modulation frequency of 1 Hz. Data was gathered over a wavelength range of 1586–1715 cm-1. With MMS all measurements were also repeated at a concentration of 1 mg/mL by further diluting the samples with the same buffer.
Results and Discussion
Robust Measurement Across a Wide Dynamic Range
Data measured for BSA across a range of concentrations (Experiment I; see Figure 2) indicate that the secondary structure of the protein is dominated by an α-helix (alpha) substructure. This substructure absorbs at a wavenumber of 1656 cm-1 and makes a fractional contribution of around 65% on average (65.29% mean, 4.23 standard deviation). Other major motifs include β-turns (turn) that absorb at a wavenumber of 1682 cm-1 and an extended structure at 1631 cm-1 (extend). These can all be robustly identified from well-documented correlations between specific wavenumbers and secondary structure motifs (4). The consistency of these results over a concentration range that spans more than three orders of magnitude sets MMS apart from techniques such as FT-IR and UV-CD that measure over a much narrower concentration window.
Figure 2: Measurements of BSA over the concentration range 0.1-200 mg/mL demonstrate the wide dynamic range of MMS, which is an important advantage for application in biotherapeutic development.
High Repeatability and Its Value in Biosimilarity Assessment
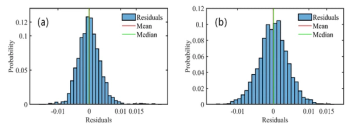
Repeat protein sample measurements were performed to demonstrate the excellent repeatability made possible by MMS. HEWL material was obtained from Sigma-Aldrich and prepared in water at a concentration of 10 mg/mL. Four common structural motifs are identified: alpha-helix ("alpha"), beta sheet ("beta"), beta turn ("turn"), and unordered structures ("unord"). At 10 mg/mL, the standard deviation of the data is around 1% for the overall structure and equally low for each motif, less than 0.5 (0.14, 0.32, 0.24, and 0.06), highlighting the ability of the technique to repeatably quantify the fractional contribution of each secondary structure motif.
Figure 3: Repeat measurements of HEWL made over the course of a month, at a concentration of 10 mg/mL demonstrate the very high repeatability of MMS, a feature that directly underpins its ability to sensitively detect structural change. (Mean and standard deviations for the fractional contribution of each motif are shown in the boxed text above each set of histograms.)
High repeatability correlates directly with the ability of a technique to detect differences, and in the case of MMS, this enables the detection of slight changes in the protein secondary structure that may have a critical impact on biotherapeutic performance. The high repeatability and reproducibility of MMS means that it can detect such differences with greater sensitivity than alternative spectroscopic techniques, such as FT-IR, and this makes it a powerful tool for similarity assessment. Figure 4 shows the set of four BSA measurements (Experiment I) displayed in the form of overlaid spectra. A number of algorithms have been proposed for the comparison of spectra for the purposes of similarity assessment, including the correlation coefficient and the area of overlap methods (8,9). Using the overlap method similarity for these samples exceeds 97% in the middle concentration range, with a drop off in similarity at both the high and low ends of the range. For replicates at 1 mg/mL, MMS typically yields area of overlap similarity values of 98.5% or higher.
Figure 4: Overlays of BSA data measured over several orders of magnitude in concentration, highlighting the value of MMS as a technique for similarity assessment throughout drug development.
This similarity figure is significantly higher than values quoted for FT-IR in the literature, which produces a mean similarity of 86.37% ± 7.98% at a single concentration of 10 mg/mL for HEWL (9). These data suggested that with FT-IR, protein similarity values comparable to those observed with the MMS data for BSA could only be obtained at significantly higher concentrations. The practical implications of this comparison are that within established biotherapeutic workflows MMS will allow more sensitive similarity assessment across a wider range of concentrations than FT-IR, a capability that is valuable both for comparability assessments during formulation development and manufacture, for example, and for biosimilarity testing in support of a submission for a follow-on biologic. A technique that generates a lower similarity for samples that are identical or exhibits deviation over the concentration range of interest, has less ability to robustly differentiate samples with genuine structural differences, compromising its value. These data also confirm the ability of MMS to generate robust measurements at far higher concentrations than UV-CD, thereby avoiding the additional workflows and dilutions that may be needed to deploy UV-CD to assess the biosimilarity of formulations at representative concentrations.
Using MMS for Protein Quantification
The measured BSA data also illustrate how MMS may be used as a protein quantification tool (Experiment I, Figure 5), more specifically the linearity of the 1656 cm-1 peak as a function of concentration over three orders of magnitude of concentration, from 0.1 to 200 mg/mL. Spectroscopy is one of a wide range of assays routinely used for protein quantification, but most spectroscopic tools have a narrow dynamic range that stems from the limited linearity of many commercial spectrometers (12). This limited linearity, which can be attributed to the combined effects of stray light, instrument slit width (resolution), and poor detector linearity, forces researchers to adjust either sample concentration or cell pathlength to achieve accurate protein quantitation. These strategies tend to be time-consuming and can directly impact data integrity.
Figure 5: Differential absorbance data measured at 1656 cm-1 shows the linearity of the MMS measurements as a function of concentration highlighting the value of the technique for protein quantification.
This linearity of the MMS data is attributable to the high resolution (<0.001 cm-1) of the instrument in combination with its low stray light susceptibility and the fact that the technique utilizes a differential measurement, thereby eliminating issues such as background subtraction that can compromise the application of conventional spectrometers. The direct control of laser power further improves linearity by managing signal dynamic range and maintaining high detector linearity across the measurement range. UV–visible (vis) methods are currently a popular choice for protein quantification, but the absorption bands in the IR region are much narrower and less dependent on aromatic residues, making MMS less susceptible to interferences. Furthermore, because MMS is not dependent on a UV chromaphore the variation of extinction coefficient is much smaller, which can be an advantage in measuring unknown proteins. Combined with a minimum measurable concentration of less than 10 µg/mL (3 sigma, HEWL) and an upper limit of greater than 200 mg/mL, these benefits make MMS significantly advantageous relative to alternative assay methods for protein quantification.
Experiment III: Comparing the Sensitivities of MMS and FT-IR in a Protein Spike Experiment
IgG1 has an almost exclusively β-strand secondary structure (86.5%) (13) while that of BSA is α-helix (94.3%) (14) (see Figure 6). Experiment III compares the relative ability of FT-IR and MMS to detect the changes that would be expected at the two wavenumbers associated with these motifs-1656 cm-1 (α-helix) and 1637 cm-1 (β-strand)-as IgG1 is spiked with BSA, at a total protein concentration of 20 mg/mL. Plotting the change in the second derivative of the FT-IR absorbance spectra, as a function of wavenumber highlights the areas of greatest change, from sample to sample, which as expected are 1656 cm-1 and 1637 cm-1 (see Figure 7a).
Figure 6: IgG1 has a secondary structure consisting predominantly of β-strands (left) whereas BSA is defined by an α-helix (right) motif. Spiking BSA into IgG1 therefore tests the sensitivity of a technique to changes in the most frequently encountered features of secondary structure.
Comparable data derived from the MMS measurements at 20 mg/mL show a much more robust linear correlation between delta second derivative data and the %BSA present (Figure 8a) than is observed with FT-IR. Sensitivity is better than the minimum experimental spike amount of 2%. These results demonstrate the superior sensitivity of MMS relative to conventional FT-IR at 20 mg/mL. Data measured by MMS at 1 mg/mL (Figure 8b) indicates a detection limit of about 4% BSA. Taken together these results indicate that MMS produces far cleaner data than FT-IR, offering up to 20 times greater sensitivity. This is a major benefit in terms of the ability to detect differences in secondary structures and therefore directly impacts the value of MMS in all measurement applications.
Figure 7: (a) FT-IR data for mixtures of BSA spiked into IgG1 indicate that the wavenumbers associated with maximum change are those associated with the α-helix and β-strand motifs, 1656 cm-1 and 1637 cm-1, respectively. (b) The technique begins to lose sensitivity to changes in the strength of the β-sheet signal at concentrations of BSA below 8%.
Conclusion
MMS is a valuable new technique for the characterization of proteins that harnesses the inherent advantages of IR spectroscopy in a configuration that is particularly well-suited to applications associated with the development, formulation, and manufacture of biotherapeutics. A key advantage is its wide dynamic range. MMS can measure over many decades of concentration, from 0.1 to 200 mg/mL (down to 0.01 mg/mL for protein quantification applications), in contrast to the single decade of concentration measurement associated with alternative techniques for measurement of the secondary structure of proteins, such as FT-IR and UV-CD. This wide dynamic range means it allows measurement at the concentration of interest from the early stages of biotherapeutic development through formulation into manufacture, with negligible requirements for sample dilution or concentration.
Figure 8: Plots of delta second derivative as a function of %BSA derived from MMS measurements (a) at 20 mg/mL and (b) at 1.0 mg/mL demonstrate the greater sensitivity of the technique relative to FT-IR.
Furthermore, MMS offers a streamlined workflow, with rapid and continuous autoreferencing that eliminates slow and inconsistent manual buffer subtraction. This characteristic results in exceptional sensitivity and repeatability, boosting its value as an analytical tool across the pharmaceutical pipeline. The recent introduction of MMS technology puts a powerful new tool in the hands of protein scientists that can be used for quantitation, the elucidation of stability and aggregation mechanisms, structural analysis, and biosimilarity assessment. Its use has the potential to streamline analyses for the commercialization of new products and bring valuable biotherapeutics to market more safely and efficiently.
References
(1) International Conference on Harmonization, ICH Harmonised Tripartite Guideline Q5E "Comparability of Biotechnological/Biological Products Subject to Changes in the Manufacturing Process" Available at:
(2) A. Elliott and E.J. Ambrose, Nature 165, 921-922 (1950).
(3) H. Fabian and W. Mantele, Handbook of Vibrational Spectroscopy , J.M. Chalmers and P.R. Griffiths, Eds. (John Wiley & Sons, Ltd., Chichester, UK, 2002), pp. 3399–3425.
(4) J.K. Koenig and D.L. Tabb, in Analytical Applications of FTIR to Molecular and Biological Systems, J.R. Durig, Ed. (D. Reidel, Boston, Massachusetts, 1980), pp. 241–255.
(5) A. Dong, P. Huang, and W.S. Caughey, Biochemistry 29, 3303–3308 (1990).
(6) A.M. Pots et al., Eur. J. Biochem. 252, 66–72 (1998).
(7) B. Shivu, S. Seshadri, J. Li, K.A. Oberg, V.N. Uversky, A.L. Fink, Biochemistry 52, 31 (2013).
(8) B.S. Kendrick, A.C. Dong, S.D. Allison, M.C. Manning, and J.F. Carpenter, J. Pharm. Sci. 85(2), 155–158 (1996).
(9) J. D'Antonio, B.M. Murphy, M.C. Manning, and W.A. Al-Azzam, J. Pharm. Sci. 101(6), 2025–2033 (2012).
(10) W. Wang and C.J. Robert, Aggregation of Therapeutic Proteins (Wiley, Hoboken, New Jersey, 2010).
(11) S.M. Kelly and N.C. Price, Current Protein and Peptide Science 1, 349–384 (2000).
(12) J.E. Noble and M.J. Bailey, Methods Enzymol. 463, 73–95 (2009). doi: 10.1016/ S0076-6879(09)63008-1.
(13) E.O. Saphire, P.W. Parren, R. Pantophlet, M.B. Zwick, G.M. Morris, P.M. Rudd, R.A. Dwek, R.L. Stanfield, D.R. Burton, and I.A. Wilson, Science 293, 1155–1159 (2001).
(14) A. Bujacz, Acta Crystallogr. D Biol. Crystallogr. 68, 1278–1289 (2012).
Eugene Ma is the Chief Technology Officer at RedShiftBio in Burlington, Massachusetts. Libo Wang is the Senior Applications Specialist at RedShiftBio. Brent Kendrick is the Vice President, Elion Labs, (Div. of KBI Biopharma) in Louisville, Colorado. Direct correspondence to:
Articles in this issue
over 7 years ago
Trends in X-ray Techniquesover 7 years ago
The C=O Bond, Part VI: Esters and the Rule of Threeover 7 years ago
Spectroscopy July 2018 Regular Issue PDFNewsletter
Get essential updates on the latest spectroscopy technologies, regulatory standards, and best practices—subscribe today to Spectroscopy.
Advertisement
Related Content
Advertisement
![Figure 3: Plots of lg[(F0-F)/F] vs. lg[Q] of ZNF191(243-368) by DNA.](https://cdn.sanity.io/images/0vv8moc6/spectroscopy/a1aa032a5c8b165ac1a84e997ece7c4311d5322d-620x432.png?w=350&fit=crop&auto=format)


Advertisement
Advertisement