News
Article
Spectroscopy
An Enhanced Approach to Analytical Instrument and System Qualification
United States Pharmacopoeia (USP) general chapter <1058> on Analytical Instrument Qualification (AIQ) now has a new title from a draft update for public comment: Analytical Instrument and System Qualification (AISQ). A new three-phase integrated lifecycle phase approach to qualification and validation is described. But will laboratories and suppliers implement it?
This column continues the discussion by Lotfinia and myself on qualifying or validating a spectrometer (1) by focusing on the recently published update of USP <1058> published in the March–April 2025 issue of Pharmacopoeial Forum (2). The “Focus on Quality” and “Questions of Quality” columns in LCGC have featured discussions of USP <1058> over the past 15 years (3–8). Although USP <1058> is for Good Manufacturing Practice (GMP) laboratories, it contains important guidance for Good Laboratory Practice (GLP) laboratories.
Terminology is important, and in the context of USP <1058>, “instrument” means any apparatus (Group A), instrument (Group B), or instrument system (Group C), used in pharmaceutical analysis. USP <1058> is an informational general chapter that provides an overall approach for the qualification and validation of instruments and instrument systems. This column does not provide a line-by-line review of the proposed update, but it discusses the most important points.
A Brief History of USP <1058>
USP <1058> is unique because it is the only pharmacopoeial general chapter providing a framework for establishing fitness for intended use of apparatus, analytical instruments, and instrument systems (9). It originated at an American Association of Pharmaceutical Scientists (AAPS) conference on analytical instrument validation in 2003, where the first agreement was that the conference name was wrong and that qualification replaced validation. At the time, the discussion was focused on addressing concerns about the overreliance on documentation, particularly for simpler pieces of laboratory equipment. A white paper was published in 2004 (10) and became the basis for USP <1058> in 2008 (11). In 2010, AAPS organized a review meeting at their annual conference where I presented a European perspective on <1058>. I introduced my presentation with a quote from that well-known qualification expert, William Shakespeare: I have come to bury <1058> not praise it. The major problems of the original USP <1058> were:
- Supplier: The true role of supplier was missing. An instrument or system is designed, built, tested, and marketed long before a laboratory considers a purchase.
- Design Qualification (DQ): Users are responsible for DQ by writing a user requirements specification of the instrument, not the supplier.
- Software Validation: There was poor software validation guidance. Software in Group B instruments implicitly validated during qualification but verification of in-built calculations or user defined programs was omitted. Validation of Group C instrument control and processing software was not a laboratory problem but the responsibility of the supplier—dream on!
For more detail, my presentation was published in this column in the same month as the conference (3).
Following this, Chris Burgess and I wrote an USP Stimulus to the Revision Process article, where Group B instruments and Group C systems were each subdivided into three types to identify what further qualification should be done (12). The aim was to integrate both instrument qualification with software validation. A risk assessment based on the three groups and the subtypes was published by us in 2013 (13), a refinement of which is in the proposed update (2). Revision of the original USP <1058> resulted in a new version in 2017 that is still current (9).
The current draft has not appeared magically out of thin air. USP published three Stimulus to the Review Process articles during 2022 (14–16), and the feedback from them has been used to prepare the update.
Proposed USP <1058> Structure
The update is a complete revision. Some parts have been reused, but there are also new sections, which are described below:
1. Introduction with an updated data quality triangle showing the impact of USP <1220> on AISQ (Partially Changed)
2. <1058> Interrelationships with Other USP General Chapters (New Section)
3. Life Cycle Process for Qualification and Validation (New Section)
4. Risk Assessment to Establish Extent of Qualification and Validation Activities (Addition and Reuse)
5. Life Cycle Models for Qualification and Validation in AISQ (New Section)
5-1 The 4Qs Model: URS and DQ, IQ, OQ, PQ (Partially Changed)
5.2 An Integrated Life Cycle Approach (New Section)
6. Calibration in Establishing “Fitness for Intended Use” of Instruments and/or Systems (New Section)
7. Change Control Over the Life Cycle (Partially Changed)
8. Roles and Responsibilities Over the Life Cycle (Partially Changed)
Overall, this is a well-structured update with more detail and figures to explain key items in the life cycle of instruments. The emphasis is on an instrument’s life cycle as a journey from specification to retirement. The headings above have life cycle mentioned five times. Unlike the current version, there is no glossary, which is a disadvantage.
<1058> and Instrumental General Chapters
Mentioned earlier, USP <1058> is an informational general chapter and outlines what needs to be done and represents best practice for AISQ. However, it does not provide any operational parameters to qualify or test. This is the role of the mandatory instrument chapters below 1000 in the USP. The draft provides a table as an example stating the mandatory and informational chapters for spectroscopic techniques where the parameters and acceptance criteria are found in the former. For many techniques, there are pairs of chapters: mandatory requirements and best practices, shown in Figure 2 of the draft document. An exception is USP <621>, which contains no chromatography qualification parameters (17,18).
Integrated Life Cycle Approach
A new three-phase life cycle approach to the qualification and validation of analytical instruments and systems is introduced in the proposed update (2). This has not been made on a whim by USP. It is an alignment based up the updated FDA Guidance for Industry on Process Validation (19). The updated FDA process validation guidance has three stages: process design, process qualification and continuous process verification, and it is shown at the bottom of Figure 1 and how it matches with a Data Integrity Model (20).


FIGURE 1: USP <1058> proposed integrated lifecycle approach aligns with process validation and analytical procedure lifecycles.
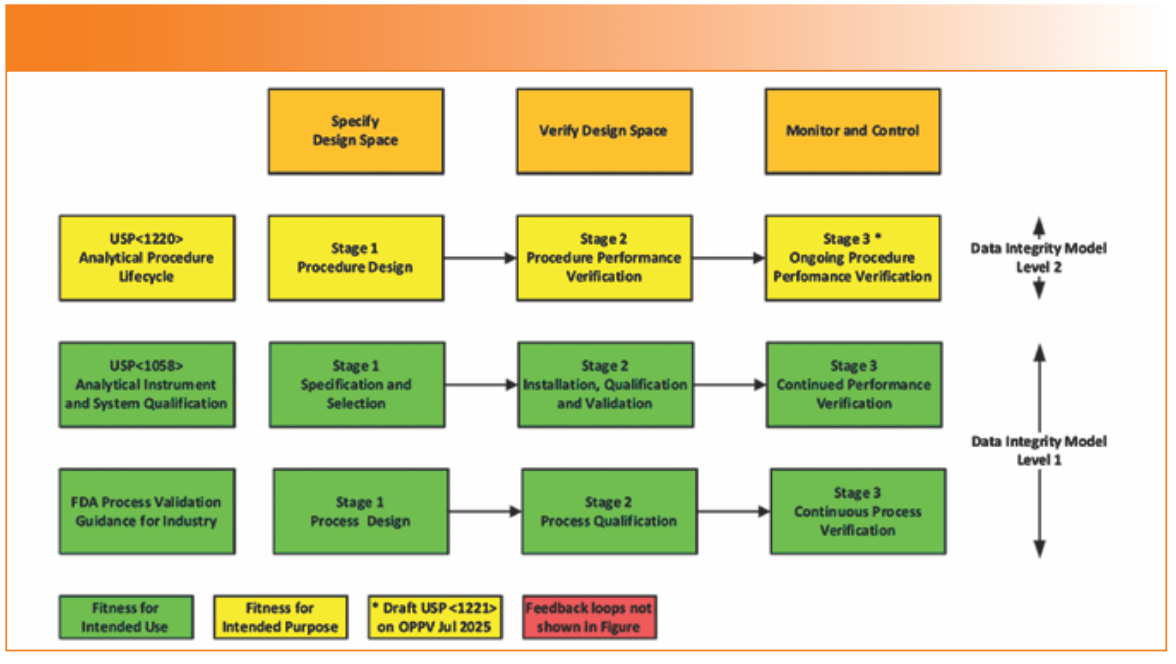
Mirroring this approach, USP <1220> on analytical procedure life cycle also has three stages: procedure design, procedure performance verification, and ongoing procedure performance verification (OPPV) (21). A draft USP <1221> on OPPV is due for publication this year to support performance of analytical procedures to assure they remain fit for their intended purpose which aligns with ICH Q14 (22).
To continue this alignment, USP <1058> has adopted a similar three-phase life cycle (2):
1. Specification and Selection
This stage covers specifying the instrument’s intended use in a URS, selection, risk assessment, supplier assessment (if applicable), and purchase.
If software is involved, a validation plan or similar controlling document is required, but there is no mention in the draft.
2. Installation, Performance Qualification, and Validation
This is an abysmal title. What performance qualification is doing here is anyone’s guess!? A much better title is “Installation, Qualification, and Validation,” and it is shown in Figures 1 and 2. This is a combination of Table I and Figure 5 in the draft document, and it illustrates that more work is required as you move from Group A to C.
In this phase, the instrument is installed and the components are integrated together and commissioned, followed by qualification, validation, or both. Qualification and validation is performed on configured software. Prior to this, there may be prototyping to confirm working practices and software configuration settings to ensure data integrity and, where necessary, operation of custom software modules. Writing SOPs and conducting user training occur here. Release for operational use is the outcome of this phase of the life cycle, and if software is involved for Group C systems, a validation summary report is missing in action.
3. Ongoing Performance Verification (OPV)
Demonstrating that the instrument performs against the requirements of the URS over the life cycle. OPV also includes maintenance, repair, calibration, service, change control, and periodic review, as appropriate. I have omitted mention of PQ from the draft.
The integrated approach was discussed briefly in our last “Focus on Quality” column where Figure 2 (1) depicts the three phases against the different 4Qs models for AIQ and CSV. Much more detail of the integrated approach can be found in the European Compliance Academy (ECA) Guide for An Integrated Approach to Analytical Instrument Qualification and System Validation (23), which is available free for members from Documents - ECA Analytical Quality Control Group (analytical-quality-control-group.org). You can join for free. The new draft Eurachem guide equipment qualification guide has been influenced by this (24).
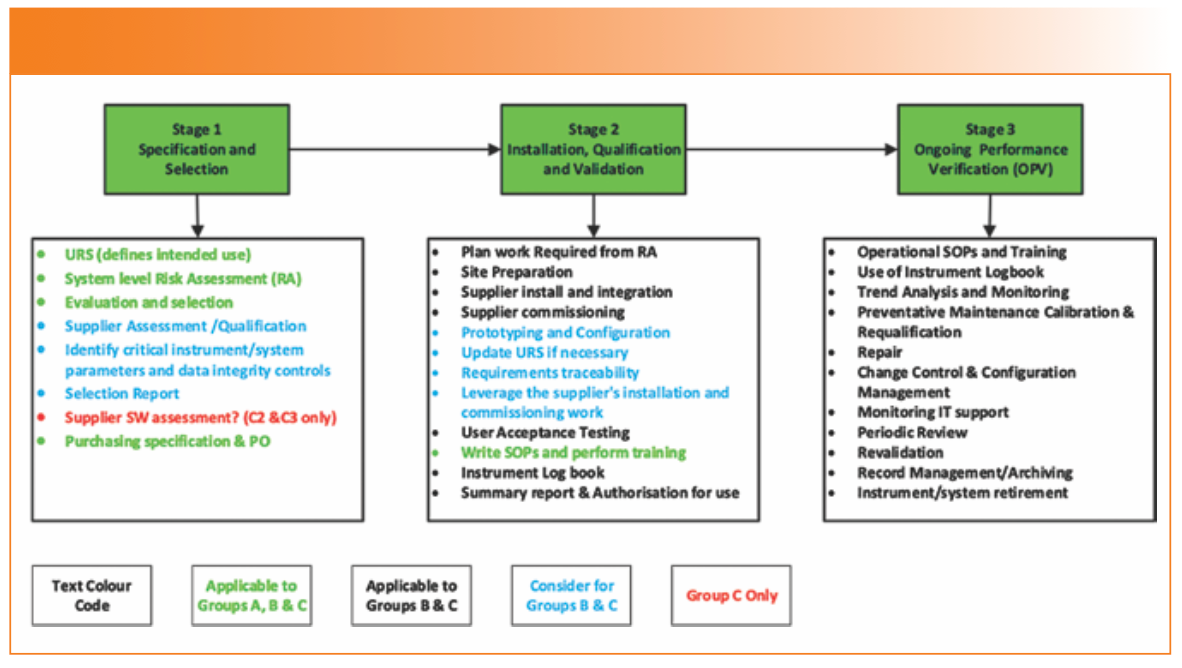
The draft USP <1058> has a figure and a table outlining the activities to be considered for each of the three phases against the three Groups and their subtypes. Figure 2 is an amalgam of both and is color-coded for common tasks and those for a specific Group. More qualification and validation work is required as you move from Group A to C, illustrating risk management in action.

.")
FIGURE 2: Activities to be performed across all groups in the integrated life cycle approach (2).
Fitness for Intended Use
What does the phrase “fit for intended use” mean for an analytical instrument and/or system as mentioned in Figure 1? The draft mentions that an instrument is under life cycle management and requires documented evidence that demonstrates that:
- It is metrologically capable of operating over the ranges specified in the analytical procedures in which it is to be used;
- Its qualification and calibration baseline, as specified in the URS, is traceable to national or international standards;
- Its overall metrological contribution to the uncertainty budget of the reportable value should be small and should preferably contribute not more than 1/3rd of the target measurement uncertainty of the analytical procedure specified in the Analytical Target Profile (ATP); and
- The instrument/system’s critical parameters continue to be in a state of procedural and statistical control and within the established acceptance limits during ongoing performance verification using a metrologically sound process over the life cycle (2).
These criteria focus solely on the instrument function and do not mention firmware or software.
Defining Intended Use
Figure 1 shows that the key to success with all three guidance and informational documents is Stage 1.
You have failed if you don’t define what you want the process, instrument, system, or procedure to do.
For USP <1058>, it is critical to define the intended use of any instrument or system to comply with the GXP regulations shown in Table I. This is achieved by writing a generic user requirements specification (URS), which must include the operating parameters and acceptance criteria defined in the mandatory instrument general chapter. You should not use tighter acceptance criteria than specified in a pharmacopoeia. The URS is a living document: as your knowledge of the instrument or instrument system increases or the intended use changes, so does the URS.



It is critical that you do not copy a supplier’s instrument specification as the parameters are measured under very tightly controlled environmental conditions that you cannot repeat in your laboratory.
Regardless of the terminology used (intended use, purpose, or appropriate design from Table I), you cannot qualify or validate an instrument or instrument system without a specification.
Updated Groups and Risk Classification
The classification of instruments into groups is a part of risk assessment in USP <1058>. The subtypes in Groups B and C are the same as in the current version, but as mentioned in a stimulus article (16), Group A is now subdivided into apparatus that either have no metrological function or no user calibration required. The Groups and subdivisions are now:
- Apparatus
A1: No metrological function
A2: No user calibration required - Instrument
B1: Firmware control
B2: In-built calculations
B3: User defined routines - System
C1: Non configurable application software
C2: Configurable application software
C3: Configurable application software and custom modules
Examples of A1 can be vortex mixers or nitrogen evaporators and A2 examples are volumetric glassware (Grade A naturally) and sieves. As always, the intended use of any instrument defines the group, so the same one can be different groups depending on intended use.
A good addition is the process flow for instrument classification that is an update of that published here in 2013 (13).
4Qs Model
Nearly a quarter of a century after the FDA dumped qualification terminology from the 2002 General Principles of Software Validation (27), GAMP 5 first edition purged it from existence in 2008 (31). Over a decade after the replacement of IQ, OQ, and PQ in the FDA Process Validation guidance of 2011 (19), the 4Qs model staggers on in EU GMP Annex 15. However, it is focused on equipment, facilities, utilities, and systems, and not on analytical instruments (32) and USP <1058>. A major problem with the 4Qs model is that there always has been a disconnect and confusion between interpretation for analytical instrument qualification and computerized system validation (1), and this is acknowledged explicitly in section 5.1 (2).
One of the difficulties associated with pharmacopeial general chapters is the balance of instructional information and options that can be interpreted. If there are too many instructions, industry will push back. This is because instrument companies, consultants, regulators, and regulated companies have differing and diverse opinions about how each life cycle stage should be addressed. As much as I want the 4Qs model to ride off into the sunset or be put out of its misery, it is ingrained with both laboratories and suppliers. Will they change and use the simpler integrated approach?
The 4Qs model has been discussed in my LCGC and Spectroscopy columns previously only the key changes in the draft will be presented here:
- The whole section on 4Qs is greatly expanded compared with the current version, with some elements having been retained and others added.
- The draft states that you can do a task to qualify an instrument, and it does not matter where it goes in the 4Qs model. One exception would be not to write the URS with acceptance criteria after the OQ has been performed.
- A URS is expected to be minimal for only A1, A2, and B1 instruments rather than all commercially available instruments as in the current version of <1058>. However, this recognizes that as an instrument’s complexity increases so should the instrument specification—compare a pH meter with an LC–MS/MS instrument, for example.
- Design Qualification (DQ) is the documented activities involved in the comparison of the laboratory URS with documentation provided by a supplier during the selection process. For Group C systems, ensure that a supplier’s quality system can produce reliable instruments, software, and network connectivity. This must be performed prior to purchase. For more complex systems, a selection report may be required to justify the purchase of a specific system.
- For a Group C instrument system, there needs to be section in the URS to define the functions of the software. This will also require a validation plan (and a corresponding summary report), which is not explicitly stated in the draft document.
- Installation Qualification (IQ) covers installation of the instrument and the integration of components such as probes and autosamplers. The verification of functionality for firmware and software for instrument control, data acquisition, storage, and retrieval then follows. Where appropriate, connection to a network for time synchronization or backup should be included.
- Operational Qualification (OQ) tests should include operating parameters and acceptance criteria from the applicable mandatory general chapter. Annex 15 clause 2.5 notes that qualification documents can be combined (32), which is also in the current version of USP <1058> but not in the update (2).
- OQ for Group C systems must be performed on configured software and, if necessary, any custom modules. Any prototyping of software configuration settings must be completed before the commencement of the OQ.
- Where appropriate storage, backup, and archive, should be undertaken to comply with applicable GXP regulations (25,28,33)
- Performance Qualification (PQ) is probably the most misunderstood portion of the 4Qs model. PQ verifies fitness for intended use under actual routine conditions of use (2,9). This demonstrates compliance with the URS.
- The user must define PQ plans including test procedures, acceptance criteria and frequency (2,9).
- PQ must not be confused with System Suitability Tests (SSTs). The draft explicitly states that SSTs may provide data on instrument performance but by itself is not PQ. This topic was discussed for HPLC in more detail by Smith and McDowall (18). Evaluate which SSTs could be used for explicit instrument testing (calibrated mass sets for balances or polystyrene standards for IR instruments) or implicit instrument testing (consistent retention time equating to HPLC pump flow rate using standard compounds and conditions).
- The relationship between the OQ and PQ stages is critical to compliant instrument use, but is perhaps the area where there is most disagreement.
Calibration
This section uses the IUPAC definition of calibration and then references other international standards for calibration (2). To bring the discussion back to pharma, it states that wavelength accuracy of a UV spectrometer between 200 and 400 nm should be ±1 nm using a certified reference standard traceable to a National Metrological Institute or standards organization (2). Traceability is one of the 10 ALCOA++ criteria for data integrity (34). Measurement uncertainty is now included in this section for the first time rather than the glossary in the current version.
This section focuses on instrument calibration performed when there is a preventative maintenance followed by a repeat qualification. The section would benefit from a discussion of the role of laboratory calibration checks and the role of the use of instrument internal calibration checks. I would recommend the referencing of question #1 of the FDA Level 2 discussion on the use of external masses versus internal instrument calibration checks for analytical balances (35).
Change Control
Changes to qualified instruments and systems must be under control. To document the potential impact of any instrument failure, the performance of the component, module, or instrument that failed must have been evaluated. In many instances, if a laboratory relies only on System Suitability Criteria for HPLC PQ, there may be no evidence to safely document the potential impact on performance.
The draft suggests only implementing changes that benefit the users or are necessary. Care needs to be taken with this approach as you could find your instrument is a candidate for your Museum of Analytical Antiquities (36). This applies, in particular, to software. Therefore, an assessment of recommended software upgrades should be performed to ensure the instrument does not have any vulnerabilities or additional risks because of not taking an upgrade.
Any change, including repair, needs to be assessed to determine if any requalification or revalidation is required to ensure that it meets the requirements in the URS and is maintained in a qualified or validated state. Software revalidation does not have much emphasis in this section, but it is just as relevant. It may be appropriate that a change triggers an update of the URS or test protocols. A configuration log is required of the instrument (make, model, serial number, and firmware revision) with similar details for the version of controlling software application, operating system, and workstation.
Roles and Responsibilities
The final section of the draft is an expanded section on roles and responsibilities. There is a figure that is a modification of the data quality triangle that integrates USP general chapters on analytical procedure life cycle, validation, and verification, plus an extension to include the roles and responsibilities of the supplier and user (2). This is an adoption of an earlier diagram (3).
USP <1058> Update: The Good, The Bad and The Ugly
To summarize the changes in the draft update, I have categorized the main changes of the proposed USP <1058> update in Table II. They are classified them as the good, the bad, and the downright ugly. This is my personal view. There are more improvements in the good section than the other two combined, which is always an advantage.



Summary
A proposal to update USP <1058> on Analytical Instrument and System Qualification was published in Pharmacopoeial Forum on March 3rd 2025. This column is my review of the changes. There is a 90-day comment period, and I would encourage all to read the proposed changes and submit your comments to USP.
In my opinion, there are many good points that will benefit all that perform integrated AISQ. If the bad and the ugly elements are removed, the final version will be an enhancement of the current version and of benefit to all working in regulated laboratories.
Acknowledgments
I would like to thank Mahboubeh Lotfinia, Jim Henderson, Eberhard Kwaitowski, and Monika Andraos and two others for reviewing this article.
References
- Lotfinia, M.; McDowall, R. D. Do We Qualify or Validate a Spectrometer? Spectroscopy 2024, 39 (8), 22–30.
- USP <1058> Analytical Instrument and System Qualification in Process Revision. Pharmacopeial Forum 2025, 51 (2).
- McDowall, R. D. Focus on Quality: Where Are We Now With USP <1058>? Spectroscopy 2010, 25 (11), 24–31.
- McDowall, R. D. USP <1058> and the GAMP Guide on Laboratory Computerized Systems – Is Integration Possible? Spectroscopy 2011, 26 (12).
- McDowall, R. D. What’s New in the New USP <1058>? LCGC Eur. 2018, 31 (1), 36–41.
- Smith, P.; McDowall, R. D. Data Integrity and USP <1058>: Part 1 Specifications and Suppliers. LCGC Eur. 2018, 31 (7), 385–389.
- Smith, P.; McDowall, R. D. Data Integrity and USP <1058>, Part 2: OQ Supervision and Execution. LCGC Eur. 2018, 31 (9), 504–511.
- Smith, P.; McDowall, R. D. Data Integrity and USP <1058>: Part 3: Monitoring and Requalification. LCGC Eur. 2019, 32 (1), 28–32.
- USP General Chapter <1058> Analytical Instrument Qualification; United States Pharmacopeia Convention Inc.: Rockville, MD.
- AAPS White Paper on Analytical Instrument Qualification; American Association of Pharmaceutical Scientists: Arlington, VA, 2004.
- USP 31 General Chapter <1058> Analytical Instrument Qualification; United States Pharmacopeia Convention Inc.: Rockville, MD, 2008.
- Burgess, C.; McDowall, R. D. Stimulus to the Revision Process: An Integrated Risk Assessment for Analytical Instruments and Systems. Pharmacopeial Forum 2012, 38 (1).
- Burgess, C.; McDowall, R. D. An Integrated Risk Assessment for Analytical Instruments and Computerised Laboratory Systems. Spectroscopy 2013, 28 (11), 21–26.
- Burgess, C.; et al. Stimulus to the Revision Process: Analytical Instrument and System (AIS) Qualification, to Support Analytical Procedure Validation over the Life Cycle. Pharmacopeial Forum 2022, 48 (1).
- Weitzel, M. L. J.; et al. Stimulus to the Revision Process: Measurement Uncertainty Evaluation Relevant to Analytical Instrument and System (AIS) Qualification - The Role of Measurement Uncertainty Concepts within the AIS. Pharmacopeial Forum 2022, 48 (2).
- Burgess, C.; et al. Stimulus to the Revision Process: Analytical Instrument and System (AIS) Qualification: The Qualification Life Cycle Process. Pharmacopeial Forum 2022, 48 (5).
- USP General Chapter <621> Chromatography; United States Pharmacopeia Convention Inc.: Rockville, MD.
- Smith, P.; McDowall, R. D. Are You Sure You Understand USP <621>? LCGC Int. 2024, 1 (8), 22–30.
- Guidance for Industry Process Validation: General Principles and Practices; Food and Drug Administration: Silver Spring, MD, 2011.
- McDowall, R. D. Understanding the Layers of a Laboratory Data Integrity Model. Spectroscopy 2016, 31 (4), 15–25.
- USP General Chapter <1220> Analytical Procedure Lifecycle; United States Pharmacopeia Convention Inc.: Rockville, MD, 2022.
- ICH Q14 Analytical Procedure Development. Step 4 Final; International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH): Geneva, 2023.
- Guide for an Integrated Lifecycle Approach to Analytical Instrument Qualification and System Validation; European Compliance Academy: Heidelberg, Germany, 2023.
- Halder, E. Draft: The Fitness for Intended Use of Analytical Equipment and Systems: A Laboratory Guide to the Lifecycle of Analytical Equipment and Systems, Their Qualification and Related Topics; Eurachem, 2025.
- 21 CFR 211 Current Good Manufacturing Practice for Finished Pharmaceutical Products; Food and Drug Administration: Silver Spring, MD, 2008.
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Chapter 3: Premises and Equipment; European Commission: Brussels, 2014.
- Guidance for Industry General Principles of Software Validation; Food and Drug Administration: Rockville, MD, 2002.
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 11: Computerised Systems; European Commission: Brussels, 2011.
- 21 CFR 58 Good Laboratory Practice for Non-Clinical Laboratory Studies; Food and Drug Administration: Washington, DC, 1978.
- OECD Principles on Good Laboratory Practice; OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring No. 1; Organisation for Economic Co-operation and Development: Paris, 1998.
- GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems, 1st ed.; International Society for Pharmaceutical Engineering: Tampa, FL, 2008.
- EudraLex - Volume 4 Good Manufacturing Practice (GMP) Guidelines, Annex 15: Qualification and Validation; European Commission: Brussels, 2015.
- OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring No. 17: Application of GLP Principles to Computerised Systems;


R D McDowall is the director of R D McDowall Limited and the editor of the “Questions of Quality” column for LCGC International, Spectroscopy’s sister magazine. ●








Newsletter
Get essential updates on the latest spectroscopy technologies, regulatory standards, and best practices—subscribe today to Spectroscopy.







