Training is essential before letting any analyst loose to work in a spectroscopy lab. But how effective is the training? Is training a one-off activity or an ongoing process?
R.C. McDowall is the principle of McDowall Consulting and director of R.D. McDowall Limited, and "Questions of Quality" column editor for LCGC Europe, Spectroscopy's sister magazine.
Training is essential before letting any analyst loose to work in a spectroscopy lab. But how effective is the training? Is training a one-off activity or an ongoing process?

We critically review the qualification and validation approaches in the World Health Organization Technical Report Series (WHO TRS) 1019 Annex 3 and its applicability to spectrometer systems.

The United States Food and Drug Administration (FDA) is using Remote Interactive Evaluations (RIE) to assess regulatory compliance, review submission material, or determine the timing of future inspections. Here, we look at some of the impacts of RIE on GxP laboratories. Although RIE is voluntary, is this an offer that you cannot refuse?

Are the integrated Analytical Instrument Qualification (AIQ) and Computerized System Validation (CSV) new approaches to the qualification and validation of laboratory systems, or are they more of the same?

The publication of a new FDA compliance policy for pre-approval inspections (PAIs) introduces a new objective to inspect research and development in pharmaceutical development. We explore how this could influence the PAI process now.

Now that the draft FDA guidance on computer software assurance (CSA) is out, we can see what it is—and is not.

A look at two ICH draft publications reveals that the EWG has much revision work to do for these publications to fully cover the validation lifecycle process.

In the first article of a two-part series, the new PIC/S guidance is discussed, with particular focus on the scope, data governance, and paper records.

“SneakerNet,” or the manual transfer of data using a disk or USB stick from one computer system to another, should be long dead, but this noncompliant transfer process still survives.
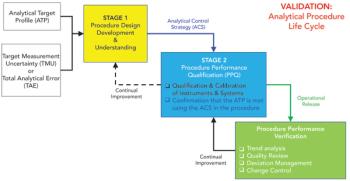
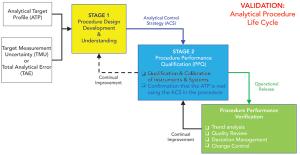
The calibration and qualification of analytical systems is a journey, not an event.

Two recent warning letters show that the US FDA is substantially increasing the amount of remediation work it requires for companies to correct data integrity noncompliance. That work can be very expensive—far exceeding the cost of ensuring compliance in the first place.

Spreadsheets are often used to perform GMP-related calculations, but can lead to serious problems and unnecessary risk. We explain why the use of spreadsheets is heavily discouraged in a regulated laboratory environment.

Spectroscopy
The Food and Drug Administration (FDA) has issued an updated version of its Compliance Policy Guide for Pre-Approval Inspections (PAI). This article will guide you on this policy’s impact on analytical procedures and data included within regulatory submissions.
Spectroscopy
Data integrity has been the subject of many “Focus on Quality” columns over the past few years (1–5). However, we have never mentioned, let alone discussed, data quality.
Spectroscopy
Analysis of FDA Form 483 observations and warning letters for infrared spectrometers reveals a range of data integrity problems and a lack of laboratory procedures for the technique. Is your laboratory in the same situation?
Spectroscopy
In the current data-integrity–centric world, is outsourcing your spectroscopy work a good idea? To answer this question, one must consider several factors.
Spectroscopy
Regulatory definitions can be confusing at all levels. In this article clear definitions of raw data are clarified and compared. A discussion of static and dynamic data is given along with a detailed summary of the impact of these terms on raw data.
Spectroscopy
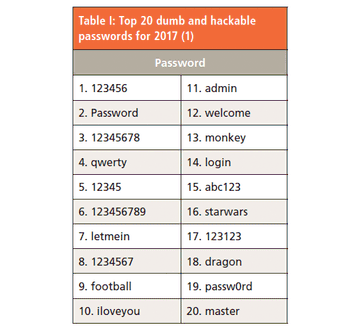
The password paradox states that simple passwords are simple to remember but easy to break, but complex passwords that are difficult to hack are also difficult to remember. Is there a better way?
Spectroscopy
Does a one size fits all data life cycle meet laboratory requirements for data integrity? No! The reason is that there are many different types of analytical procedures and this requires a flexible analytical data life cycle.
Spectroscopy
We live in a new world where users must specify what they want their analytical instruments to do. This discussion will help you figure out how to make sure you get what you need.
Spectroscopy
Data governance requires a multi-layered approach that runs throughout a regulated organization from top to bottom. Although data governance features in the majority of GxP data integrity guidance documents, the approach to the topic should be business-driven rather than regulatory-driven.
Spectroscopy
Raw data is a term that is often used in both good manufacturing practice (GMP) and good laboratory practice (GLP) laboratories but can create confusion and misunderstanding. What exactly does it mean and what records are within the scope of the term?
Spectroscopy
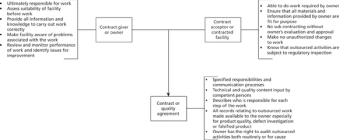
In pharmaceutical contract manufacturing, the work of analytical scientists is covered by a quality agreement, which is prepared by personnel in the quality control or quality assurance department and focuses on the laboratory analyses provided. But what exactly are quality agreements, why do we need them, who should be involved in writing them, and what should they contain? Here, we answer those questions.

September 1st 2020

May 8th 2024

September 1st 2023

December 1st 2023