Article
Special Issues
Spectroscopy Supplements
Identification of a Low-Abundant Biomarker in Human Serum Using nanoLC–LIT-TOF MS and Information-Based Acquisition Techniques
Author(s):
Serum protein profiling using mass spectrometry (MS) is one of the most promising approaches for biomarker identification. The authors adopted a nano liquid chromatography (nLC)–linear ion trap time-of-flight (LIT-TOF) MS system and newly developed software known as information-based acquisition (IBA) to identify biomarkers in human serum. IBA is a data processing protocol for repetitive MS analyses. Peptides selected for the first-pass MS-MS analysis are automatically excluded from the MS spectrum such that subsequent MS-MS analyses are performed on different peptides to minimize overlapping analyses, resulting in the identification of relatively low-abundant peptides.
Serum samples from a healthy individual containing a known biomarker (C-reactive protein) at a typical concentration reported in patients were analyzed using nano liquid chromatography linear ion trap time-of-flight mass spectrometry (LC–LIT-TOF MS) with information-based acquisition (IBA). The low-abundant biomarker in the patient serum was not detected by single MS analysis, but it was identified in the following run using the IBA function. These results suggested that IBA would be effective for the identification of low-abundant proteins and would become a powerful tool for the biomarker discovery.
The comparison of the protein expression between healthy individuals and patients is one of the promising tools for biomarker identification. MS coupled to LC separation has been used widely for the analyses of these complicated biological samples. Although there have been significant technical improvements in these methodologies, some of the challenges, such as the detection of low-abundant proteins, remain (1).
In this study, a hybrid nanoLC–LIT-TOF MS system was used along with the IBA data processing protocol for the identification of a low-abundant biomarker. With an ever-increasing demand for MS-based biomarker discovery and validation, it is crucial for MS platforms to provide high throughput solutions, high mass accuracy, increased sensitivity, and most importantly, sound quantitative analysis. Achieving this level of quantitative performance requires powerful software and sufficient dynamic range.

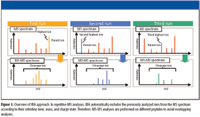
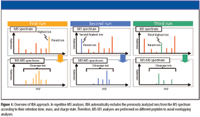
Figure 1
IBA is a data processing program for repetitive MS analyses that can record the retention time, mass, charge state, and MS-MS data in an internal database in real time. In subsequent runs of the same sample, MS-MS analyses are performed on different peptides to minimize overlapping analyses. This enhanced MS-MS efficiency allows the identification of relatively low-abundant proteins. An overview of IBA is shown in Figure 1. Here, we compare the protein components of serum from a healthy individual with serum containing a known biomarker doped in at a concentration consistent with that reported in patients, using the IBA data processing program. The biomarker was successfully identified, suggesting that IBA would be a powerful tool for further proteomics analyses, especially for the investigation of additional low-abundant biomarkers.
Experimental
As a model biomarker, C-reactive protein (CRP) was adapted for this study. CRP is a known indicator of inflammation diseases like bacterial infection and collagen diseases, which are associated with marked serum CRP elevation (more than 0.1 mg/mL) in their acute phase. In this study, the patient serum was prepared by adding CRP (30-AC05, Fitzgerald, Concord, Massachusetts) at a concentration of 0.1 mg/mL to the commercially obtained human serum (H1388, Sigma, St. Louis, Missouri).
Six high-abundant proteins — albumin, IgG, IgA, transferrin, antitrypsin, and haptoglobin — were depleted using a Multiple Affinity Removal column (5185-5984, Agilent Technologies, Wilmington, Delaware). The obtained samples were dissolved in 50 mM ammonium bicarbonate containing 8 M urea, 2.5 mM EDTA, and 10 mM dithiothreitol, and incubated at 37 °C for 1.5 h for reduction. Iodoacetamide was added at a concentration of 50 mM and incubated at room temperature for 0.5 h for alkylation. After removing urea by dialysis against 50 mM ammonium bicarbonate, trypsin (11 047 841 001, Roche, Clifton, New Jersey) was added at the amount of 1/50 of the protein, and incubated at 37 °C for 16 h.

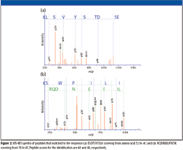
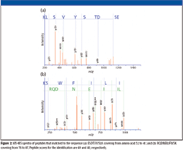
Figure 2
LC–MS analysis was performed using a NanoFrontier nanoLC–LIT-TOF MS system (Hitachi High-Technologies). The analytical conditions were as follows:
Separation column: PicoFrit (500 mm × 0.075 mm, New Objective, Woburn, Massachusetts); mobile phase: (A) 2% acetonitrile containing 0.1% formic acid, (B) 98% acetonitrile containing 0.1% formic acid; gradient: A:B = 98:2 (0 min), 60:40 (60 min), 0:100 (70–80 min); flow rate: 100 nL/min; spray potential: 1400 V, AP1 temperature: 140 °C; curtain gas flow: 1.0 L/min; mass scan range: m/z 100–2000; detector voltage: 2250 V.
IBA cycles were repeated three times with the parameter of retention time and mass tolerance under 10% and ±0.1 Da, respectively. MS-MS data were searched using MASCOT software (Matrix Science, Boston, Massachusetts) by the following conditions:
Database: NCBInr; enzyme: trypsin; missed cleavage: 1; fixed modification: carbamidomethyl; protein mass: no restriction; peptide mass tolerance: ± 0.3 Da; fragment mass tolerance: ± 0.1 Da.
Results
First, single MS analysis was performed on both the normal and the patient sera. Although more than 40 proteins were identified with more than two peptides and with MASCOT scores of MS-MS spectra higher than 30, the target CRP could not be detected. Because the dynamic range of the serum proteome spans 12 orders of magnitude (2), many other high-abundant proteins, in addition to the depleted six major proteins, were expected. Most of the identified proteins were these high-abundant classical plasma proteins (2). Therefore, MS-MS analyses were performed preferentially for the peptides derived from them, resulting in the poor identification of low-abundant proteins.
Next, three repetitive MS analyses using an IBA protocol were performed for the same sample. CRP was successfully identified at the second cycle by two peptides. The MS-MS spectra of these peptides are shown in Figure 1. The ion scores of these peptides were 40 and 49, respectively, and both were assigned to rank 1. Therefore, confidence of the identification level is high, suggesting that IBA function is useful for the identification of low-abundant proteins.
Conclusion
The splitless nano flow rates generated by nanoLC in combination with the hybrid LIT-TOF system and intelligent software offers a unique combined approach to increasing reproducibility, sensitivity, and mass accuracy, which should also lead to an increase in data quality and confidence.
This study demonstrates that repetitive MS analyses using the MS system's IBA function are effective in identifying the low-abundant proteins in a complicated biological sample that otherwise could not be identified by a single MS analysis. These results establish that the IBA function could become a powerful tool for further proteomics analyses, especially for biomarker discovery.
Yukie Sasakura, Makoto Nogami, Shinji Nagai, Susumu Watanabe, and Katsuhiro Kanda are with Hitachi High-Technologies, Hitachinaka, Ibaraki, Japan.
Jennifer Krone and Chad Ostrander are with Hitachi High-Technologies America, San Jose, California.
References
(1) A. Vlahou and M. Fountoulakis, J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 814, 11–19 (2005).
(2) N.L. Anderson and N.G. Anderson, Mol. Cell. Proteomics 1, 845–867 (2002).






Newsletter
Get essential updates on the latest spectroscopy technologies, regulatory standards, and best practices—subscribe today to Spectroscopy.










