Article
Spectroscopy
Spectroscopy
Improving Accuracy in Inductively Coupled Plasma–Quadrupole Mass Spectrometry: The Interference Standard Method
An approach for dealing with one of the main limitations of ICP-MS, low sensitivity and accuracy caused by spectral interferences
One of the main limitations with inductively coupled plasma–mass spectrometry (ICP-MS) is related to the occurrence of spectral interferences. Considering the relatively low resolution presented by quadrupole-based mass spectrometry (ICP-QMS), sensitivity and accuracy can be compromised by interfering ions formed in the plasma. Several strategies have been proposed to overcome the problem, and the most successful ones are based on high-resolution instruments, mixed-gas plasmas, or collision-reaction cells and interfaces. A different approach was recently proposed based on the idea that interfering polyatomic ions and some plasma naturally occurring Ar species present similar behaviors. The so-called interference standard method (IFS) uses the analytical to IFS signal ratio associated with external calibration to minimize the interfering species contribution to the analytical signal, thereby improving ICP-QMS accuracy. In this work, the efficiency of the IFS method in the determination of Si is evaluated. A 130% recovery was found for 28 Si determined in a standard reference material (Typical Diet, NIST 1848a) using the conventional external calibration method without any interference correction. Such high recovery is a result of spectral interferences caused by molecular ions such as N2+ and 12 C16 O+ . On the other hand, no statistically significant difference at a 95% confidence level was found between reference- and IFS-determined values using the 36 ArH+ or 38 Ar+ probes. Limits of detection of 6.0, 5.0, and 8.0 µg/L were calculated for determinations at m/z 28 or using the 28/37 and 28/38 signal ratios, respectively. Possible mechanisms responsible for the IFS method efficiency also are discussed here.
Inductively coupled plasma–mass spectrometry (ICP-MS) is a powerful instrumental technique that has become increasingly popular in recent years, especially because of a reduction in costs and the growing demand for fast, sensitive analytical methods from developing economies (1). Despite their high sensitivity and multielement capabilities, quadrupole-based instruments present an important limitation related to their intrinsic inability to solve spectral interferences in the 1 amu range. Thus, polyatomic ions with mass-to-charge ratios (m/z) close to the analytes can severely compromise ICP-quadrupole mass spectrometry (QMS) sensitivity and accuracy. Such interfering ions can be formed by interactions among species originated in the plasma, the solvent, the sample matrix, and atmosphere gases diffusing into the plasma. The problem is even more critical for species present in high concentrations in one of the interference sources — for example, 40 Ar+, 40 Ar35 Cl+, 12 C16 O+, 14 N2+, and 16 O2+ — and can compromise the determination of important elements such as Ca, As, Si, and S. Considering all of these variables, it is clear that correcting spectral interferences in ICP-QMS is not trivial. Several strategies have been proposed to overcome this problem, and the most common are based on collision-reaction cells and interfaces (2,3), mixed-gas plasmas (4,5) and mathematical correction (6,7).
A different approach to reducing spectral interferences in ICP-QMS was recently proposed by Donati and colleagues (8). The interference standard method (IFS) uses Ar species naturally present in the plasma to reduce the contribution of interfering polyatomic ions to the analytical signal. This method uses the same reasoning behind a conventional internal standard method, but targets the interfering species rather than the analytes. The method assumes that Ar ions and some polyatomic species present similar behaviors in the plasma and that by using the analytical to IFS signal ratio associated with an external calibration method, it is possible to improve accuracy in ICP-QMS determinations. In fact, significant improvements have been reported by using this strategy even for some severely affected elements, such as S and Fe (9).
In this article, the IFS method capabilities are demonstrated by determining Si in a standard reference material and comparing the results with values obtained with a conventional external calibration method without any interference correction. Determining this element by ICP-QMS is not a trivial task, especially for nitric acid–digested samples that contain high concentrations of elements that are precursors of Si main interferences. Ions such as 14 N2+ and 12 C16 O+, which present m/z close to the most abundant isotope of Si (28 Si, 91.23%), can significantly compromise accuracy and prevent determinations in complex matrices. The IFS probes 36 ArH+ and 38 Ar+ were used to improve accuracy. Possible mechanisms contributing to the method efficiency also are discussed.
Experimental
Instrumentation
An inductively coupled plasma–quadrupole mass spectrometer (model 820-MS, Varian) was used in all determinations. The sample introduction system comprised an automatic sampler (SPS3, Varian), a concentric nebulizer, and a double-pass, Scott-type spray chamber. The spray chamber temperature was controlled by a Peltier device and was kept at 2 °C to minimize the formation of oxides. The analyte was monitored at m/z = 28, and the IFS probes were monitored at m/z = 37 and 38. Table I presents the operational conditions used in this work.

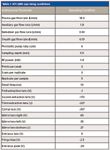
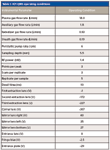
Table I: ICP-QMS operating conditions
A cavity microwave oven (Ethos 1600, Milestone-MLS) was used for sample digestion.
Reagents, Standard Reference Solutions, and Samples
Ultrapure HNO3 was produced using a sub-boiling distillation system (Milestone) and was used in the sample digestion and preparation of all standard reference solutions. Hydrogen peroxide (30% m/m, Labsynth) also was used for sample preparation. All standard reference solutions were prepared by dilution of a 1000-mg/L Si stock solution (Quemis) with distilled–deionized water (18.2 MΩ·cm, Milli-Q, Millipore) in 1% (v/v) HNO3 medium. The external calibration method was used in all determinations. All glass, propylene, or PTFE–PFA materials were kept in 10% (v/v) HNO3 overnight and rinsed with distilled–deionized water before use. The Ar plasma source was a 99.999% liquid argon Dewar (White Martins). A standard reference material (Typical Diet, SRM 1848a) from the National Institute of Standards and Technology (NIST) was used to check the method accuracy.
Sample Preparation
Approximately 250 mg of Typical Diet was accurately weighted in PTFE–PFA digestion flasks. Aliquots of 2.5 mL of concentrated ultrapure HNO3 (14 mol/L) were added to each sample replicate (n = 3) and a predigestion period of 30 min at room temperature was observed. Then, a volume of 2.5 mL of distilled–deionized water was added to each flask and an additional period of 30 min without heating was observed. Finally, 3.0 mL of H2O2 30% m/m was added to the digestion flasks, which were then submitted to microwave-assisted digestion in a cavity oven. Analytical blanks were prepared in the same manner without any sample. The digestion heating program used consisted of five steps: 250 W for 2 min at 80 °C; 0 W for 3 min at 70 °C; 550 W for 4 min at 120 °C; 650 W for 5 min at 180 °C; and 750 W for 3 min at 200 °C. The solutions were allowed to cool down, transferred to 15-mL polyethylene tubes, and diluted to a final volume of 10 mL with distilled–deionized water. Further dilution was then carried out just before analysis to ensure the analyte concentration compatibility with the analytical calibration curve linear range.
Results and Discussion
Improving ICP-QMS Accuracy
As previously discussed, the relatively low resolution of ICP-QMS may be an important limitation for complex matrix applications. This aspect is even more critical for elements presenting major isotopes that are prone to severe spectral interferences (10). Silicon's most abundant isotope at m/z 28 was used to determine this element in a standard reference material and the results for a conventional external calibration with or without applying the IFS method are compared in Table II. For the IFS method, the ratio between signal intensities at m/z 28 (analytical signal) and m/z 37 or 38 (IFS) were used for all reference solutions, blanks, and sample replicates.
As we expected, for an organic complex matrix submitted to acid digestion with HNO3, overestimated values were obtained for Si determined at m/z 28, because of spectral interferences caused by 14 N2+ and 12 C16 O+ (11). Using the external calibration method without IFS correction, a recovery of 130% was obtained for Typical Diet (NIST 1848a). On the other hand, significant accuracy improvements were observed for determinations using either the 36 ArH+ or the 38 Ar+ IFS probes. Results for the 28/37 and 28/38 ratios presented no statistically significant difference from the reference value at a 95% confidence level (Table II).



Table II: Determination of Si in typical diet (NIST SRM 1848a) by ICP-QMS using the external calibration method with or without the IFS correction
IFS Correction
The IFS method is based on the hypothesis that interfering ions and IFS probes present similar behaviors in the plasma (8). A piece of evidence to support this assumption is presented in Figure 1. In this case, a 1% (v/v) HNO3 solution was analyzed by ICP-QMS, and the signals at m/z 28, 37, and 38 were monitored during 20 consecutive measurements. In general, it can be observed that there is a similar signal profile for all m/z monitored, which may indicate similar behaviors for 14 N2+, 36 ArH+, and 38 Ar+ in the plasma. The correlation coefficients calculated from the data presented in Figure 1 were equal to 0.85 and 0.54 for the 28/37 and 28/38 pairs. The values and results presented in Table II suggest that the closer the relationship between the interfering ion and the IFS probe, the better the accuracy while using the IFS strategy. It is important to note in this case that variations in interfering and IFS signals must not only present a relatively linear relationship (that is, a correlation coefficient close to 1), but also have similar magnitudes, which may be the case considering the results in Table II. Another important observation is that an eventual signal overlap at the IFS probe m/z can compromise its efficiency. The IFS probe at m/z 37, for example, will probably have a negative effect on accuracy while analyzing samples with high concentrations of Cl because of the signal overlap from the 37 Cl isotope. In this context, an interesting advantage of using the 38 Ar+ IFS probe is that it presents no signal overlap from other species.

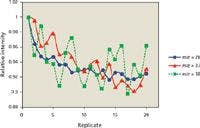
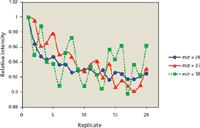
Figure 1: Signal profiles for a 1% (v/v) solution of HNO3 determined by ICP-QMS in 20 consecutive measurements.
The mechanism responsible for the IFS method efficiency is not yet understood, but it might be related to thermodynamic equilibria in the plasma and to direct and indirect interactions between interfering ions and IFS probes (8). Considering 28 Si+ spectral interferences, the possibility of direct interactions between the 36 ArH+ IFS probe and interfering precursor species such as N2 and CO may be demonstrated by the reactions represented in equations 1 and 2 (12). In this case, a decrease in the IFS signal would be indirectly related to the same behavior for the interfering species, since IFS and interfering ion precursors would be consumed. These reactions are both relatively fast and thermodynamically favorable, with ΔH equal to -1026 and -581 kJ/mol, respectively (13).

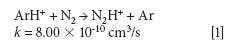
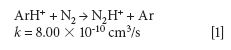
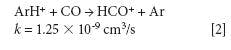
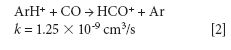
where k is a rate constant. In addition to these direct reactions, it is possible that both interfering species and IFS probes interact in a similar manner with the same reactant, which would also result in similar signal profiles. This fact is exemplified by equations 3–5 (14–16). It can be observed that the interfering ion N2+ and the IFS probes 36 ArH+ and 38 Ar+ present similar rate constant values (k) for reactions with H2O. They also present similar energy variations, that is, -288, -303, and -315 kJ/mol, respectively (13).

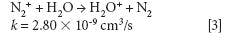

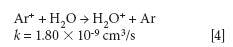

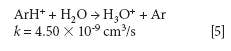
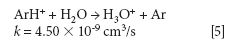
Analytical Figures of Merit
As it was observed in previous works (8,9), the IFS method has limited impact on sensitivity (Table III). This fact is expected considering that the interfering species are not physically destroyed. On the other hand, a precision depreciation would also be expected by applying the IFS method because another source of noise is being added (the IFS signal). However, depending on the source of noise, this precision depreciation could be negligible, as can be seen in Table II. The precision varies from 14% without the IFS method to 10% and 17% with the 28/37 and 28/38 ratios, respectively. Table III presents the instrumental limits of detection (LOD) and quantification (LOQ) for determinations with or without the IFS method. The LOD was calculated as three times the background equivalent concentration (BEC) and multiplied by the blank relative standard deviation (RSD, n = 20) (17). The BEC was obtained by dividing the Si concentration in one of the calibration curve reference solutions by its respective signal-to-background ratio (SBR). The SBR is the net analytical signal divided by the blank signal. The LOQ was calculated as 10 times BEC, multiplied by the blank RSD (n = 10).



Table III: Instrumental limits of detection and quantification for the determination of 28Si+ by ICP-QMS using the external calibration method with or without the IFS correction
Conclusions
The interference standard method is an interesting alternative to correct spectral interferences in ICP-QMS determinations. No instrumental modification, reagent addition, or introduction of reactive gases is required, which contributes to easy implementation in routine procedures.
It is important to note that the accuracy improvements observed in this and previous works using the IFS method may be a result of complex mechanisms. This work's goal is not to elucidate such mechanisms, but to present some pieces of evidence that may contribute to a better understanding of the method. More fundamental studies and application to different analytical contexts are necessary to access the full potential and shortcomings of the IFS strategy.
Acknowledgments
The authors would like to thank the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for grants and fellowships provided (2006/59083-9, 2010/50238-5, and 2010/17387-7). The support from the Instituto Nacional de Ciências e Tecnologias Analíticas Avançadas, Conselho Nacional de Desenvolvimento Ciências e Tecnologias (INCTAA, CNPq, and FAPESP) also is greatly appreciated.
References
(1) L.S. Schmid, Spectroscopy 25 (3), 40–45 (2010).
(2) S. D'Ilio, N. Violante, C. Majorani, and F. Petrucci, Anal. Chim. Acta 698, 6–13 (2011).
(3) C.D. Pereira, E.E. Garcia, F.V. Silva, A.R.A. Nogueira, and J.A. Nóbrega, J. Anal. At. Spectrom. 25, 1763–1768 (2010).
(4) X. Jin, L. Li, W. Hang, J. Chen, and B. Huang, Spectrochim. Acta 65B, 1052–1055 (2010).
(5) C. Agatemor and D. Beauchemin, Spectrochim. Acta 66B, 1–11 (2011).
(6) U.S. Environmental Protection Agency, Method 2060a (SW 846), 1998. http://www.epa.gov/sam/pdfs/EPA-6020a.pdf. Accessed July 19, 2011.
(7) K. Neubauer, Spectroscopy 25 (11), 30–36 (2010).
(8) G.L. Donati, R.S. Amais, and J.A. Nóbrega, J. Anal. At. Spectrom. 26, 1827–1832 (2011).
(9) R.S. Amais, G.L. Donati, and J.A. Nóbrega, Anal. Chim. Acta 706, 223–228 (2011).
(10) J.S. Becker, Inorganic Mass Spectrometry: Principles and Applications (Wiley, Chichester, UK, 2008), p. 514.
(11) P. Pohl, N. Vorapalawut, B. Bouyssiere, and R. Lobinski, J. Anal. At. Spectrom. 25, 1461–1466 (2010).
(12) H. Villinger, J.H. Futtrel, F. Howorka, N. Duric, and W. Lindiger, J. Chem. Phys. 76, 3529–3534 (1982).
(13) National Institute of Standards and Technology, Chemistry WebBook, NIST Standard Reference Database. http://webbook.nist.gov/chemistry/form-ser.html. Accessed July 18, 2011.
(14) D. Smith, N.G. Adams, and T.M. Miller, J. Chem. Phys. 69, 308–318 (1978).
(15) G. Mauclaire, R. Derai, and R. Marx, Dyn. Mass Spectrom. 5, 139–145 (1978).
(16) W. Lindinger, Phys. Rev. 7A, 328–333 (1973).
(17) V. Thomsen, D. Schatzlein, and D. Mercuro, Spectroscopy 18 (12), 112–114 (2003).
George L. Donati, Renata S. Amais, and Joaquim A. Nóbrega are with the Group of Applied Instrumental Analysis, in the Department of Chemistry at Federal University of São Carlos in São Carlos, SP, Brazil. Please direct correspondence to: georgedonati@yahoo.com.br.





Newsletter
Get essential updates on the latest spectroscopy technologies, regulatory standards, and best practices—subscribe today to Spectroscopy.








