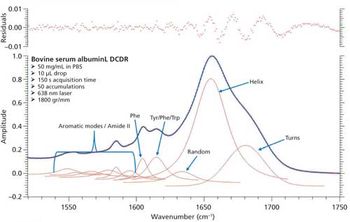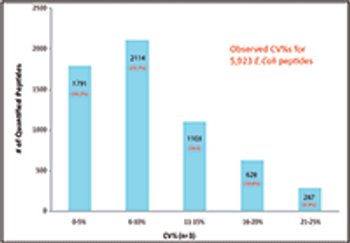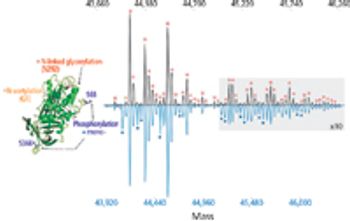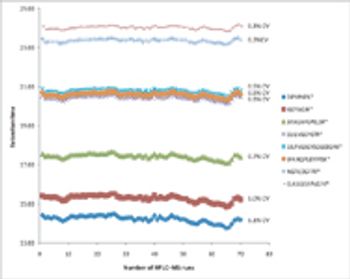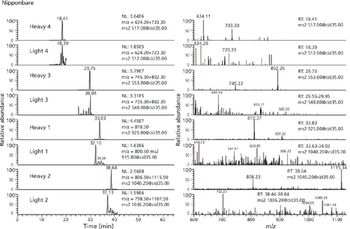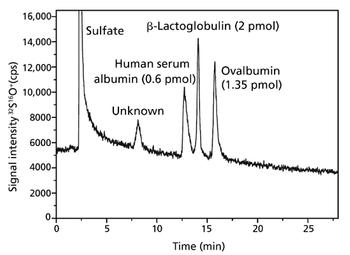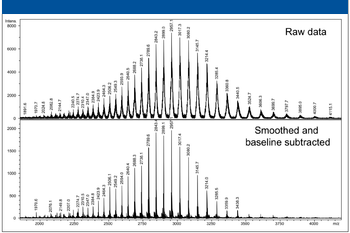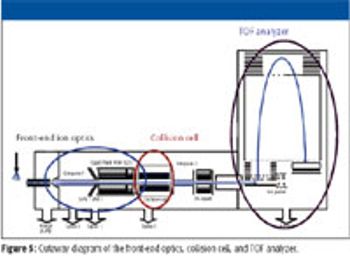

Proteomics and structural biology require specialized mass spectrometry methods for characterizing protein structures and conformations. Jennifer S. Brodbelt, a professor of chemistry at the University of Texas at Austin, focuses on the development and application of photodissociation mass spectrometry for studying biological molecules such as peptides, proteins, nucleic acids, oligosaccharides, and lipids. She recently spoke with Spectroscopy about her work with this technique. She is the winner of the 2017 ANACHEM Award, which will be presented at the SciX meeting in October 2017. The award is presented annually to an outstanding analytical chemist based on activities in teaching, research, administration, or other activities that have advanced the art and science of the field.